|
Overview
The methods and topics of life science research have changed profoundly in the last decade. Genomic science has shifted our attention from hypothesis-driven studies of individual molecules to phenomenological approaches that target comprehensive views of the living cell and its dynamics. Proteomics is setting its sights on a description of the entire complement of cellular components and their interactions.
This development has been technology driven to a large extent; as a corollary we see that the applications domain, especially the biotechnology industry, contributes an unusually large fraction of its momentum. We - as basic scientists in the traditional centres of collaboration, education and non-directed discovery - are challenged not to fall behind: experience shows that sustained progress in the private sector critically depends on the academic sector to provide an uninterrupted stream of insight and innovations in the long-term.
With this in mind, our program of biomolecular engineering research emphasizes the investigation of engineering principles and strategies that may contribute to an understanding of biomolecular systems at a level that goes beyond phenomenological descriptions and short-term application issues. Our topics span bioinformatics, protein engineering and biomolecular nanotechnology. At the core of the program is the analysis and integration of genomics and proteomics data to guide protein engineering, the experimental validation of our predictions, and the application of results for molecular medicine and bio-nanotechnology in a mid- to long-term timeframe.
The cohesive element in our research projects ist the quest to understand complexity in biomolecular systems. Complexity arises from a context dependent behaviour of system components and we observe complexity in many hierarchical layers of structure formation and generation of function, from the genome to the living cell. We focus our work mainly on proteins since protein folding is the quintessential paradigm of self-organising molecular systems. Based on our concepts to address complexity, we develop strategies and algorithms to analyse proteins and engineer them in predictable ways. We apply our understanding to interesting model proteins and biomolecular assemblies and we aim to spin out successful concepts to biotechnology and medicine.
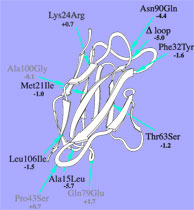
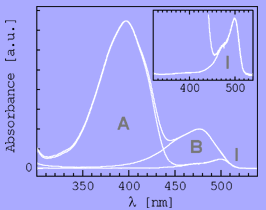
"Engineering" implies the rational application of well understood principles with predicted outcome. Since structured biomolecules are complex systems, the effects of changes are difficult to predict. "Protein Engineering" thus may be regarded to be an oxymoron, a contradiction in terms. Nevertheless, we have been able to provide at least some rationality to the engineering of antibody domains and to the designed construction of biomolecular nanoassemblies. We anticipate further progress in the application of our strategies.
|
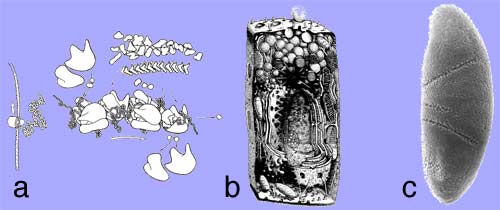
Layers of biological complexity:
(a) Protein sequences are represented in the genome, but transcriptional regulation already provides the first unknown, as to which genes are being transcribed under which circumstances. As the transcript is further processed from left to right in this schematic, regulated alternative splicing will significantly change the m-RNA message of most genes. Finally, after translating the protein, the polypeptide folds to a strcuture that cannot be predicted from knowledge of the sequence information. Thus while cellular behavior as a result of protein function as a result of protein structure as a result of protein sequence is wholly represented in the genome, we have no way of assembling the latter from the former, unless the entire context is completely specified at the same time. This is a little like having to know the answer, in order to be able to ask a question. (b) The problem of hierarchical layers of complexity, where one layer cannot be defined before the entire underlying layer is specified, is complicated further when individual proteins assemble structurally into functional molecular machines, or functionally into metabolic or signalling pathways, and other higher-order cellular subsystems. Dynamic switching by posttranslational modifications becomes an issue, and protein sorting into different compartments must be taken into account, among many other aspects. (c) No less complex, and no more predictable from first principles, is the self-assembly of cells into organs and organisms during development.
Yet the entire process is self-organized, robust and reproducible.
|
|

 The
The