Difference between revisions of "CSB Web tools"
m |
m |
||
| (11 intermediate revisions by the same user not shown) | |||
| Line 3: | Line 3: | ||
CSB on the Web | CSB on the Web | ||
</div> | </div> | ||
| − | |||
| − | |||
| − | |||
| Line 13: | Line 10: | ||
__TOC__ | __TOC__ | ||
| + | |||
| + | ==Introductory reading== | ||
| + | <section begin=reading /> | ||
| + | {{#pmid:21863497}} | ||
| + | <section end=reading /> | ||
==Contents== | ==Contents== | ||
| − | |||
| − | == | + | ===Databases=== |
| − | + | {{#pmid:20221911}} | |
| − | + | {{#pmid: 18287689}} | |
| + | ===Web servers=== | ||
| + | {{#pmid: 17993673}} | ||
| + | {{#pmid:20221929}} | ||
==Exercises== | ==Exercises== | ||
| Line 34: | Line 38: | ||
==Further reading and resources== | ==Further reading and resources== | ||
| + | {{#pmid: 24234005}} | ||
| + | {{#pmid: 24165881}} | ||
| + | {{WWW|WWW Bioinformatics.ca links}} | ||
| + | {{#pmid:22139927}} | ||
| + | {{#pmid:22086951}} | ||
| + | {{#pmid:22102583}} | ||
| + | {{WWW|WWW NAR database}} | ||
| + | {{WWW|WWW NAR webserver}} | ||
| + | {{WWW|WWW NCBI gene}} | ||
| + | {{#pmid: 18287687}} | ||
| + | {{#pmid:20221912}} | ||
| + | {{WWW|WWW UniProt}} | ||
| + | {{#pmid:20221914}} | ||
| + | {{#pmid:21225378}} | ||
| + | {{WWW|WWW SGD}} | ||
| + | {{WWW|WWW MGI}} | ||
| + | {{WWW|WWW GO}} | ||
| + | {{WWW|WWW Wiki gene}} | ||
| + | {{WWW|WWW GPSDB}} | ||
| + | {{WWW|WWW HGNC}} | ||
| + | {{#pmid:20221918}} | ||
| + | {{#pmid:20221917}} | ||
| + | {{#pmid:20221916}} | ||
| + | {{WWW|WWW Reactome}} | ||
| + | {{WWW|WWW KEGG}} | ||
| + | {{WWW|WWW BioCyc}} | ||
| + | {{WWW|WWW GMOD}} | ||
| − | + | | |
[[Category:Computational_Systems_Biology]] | [[Category:Computational_Systems_Biology]] | ||
</div> | </div> | ||
Latest revision as of 14:18, 8 January 2015
CSB on the Web
Important tools and resources for CSB, available on the Web.
Contents
Introductory reading
| Kowald & Wierling (2011) Standards, tools, and databases for the analysis of yeast 'omics data. Methods Mol Biol 759:345-65. (pmid: 21863497) |
|
[ PubMed ] [ DOI ] One of the major objectives of systems biology is the development of mathematical models for the quantitative description of complex biological systems, such as living cells. Biological data and software tools for the design, analysis, and simulation of models are two basic ingredients for the new field of systems biology. In this chapter we give an overview of databases and repositories that provide valuable information for the integrative analysis and modeling of data generated by the different omics techniques. We also provide a review of the most popular software tools currently used in computational systems biology studies. Standards for the annotation of biological data and for the analysis and exchange of models are fundamental for the success of systems biology and provide the glue that connects experimental data with mathematical models. We also discuss some broad trends regarding where systems biology is heading to. |
Contents
Databases
| Tatusova (2010) Genomic databases and resources at the National Center for Biotechnology Information. Methods Mol Biol 609:17-44. (pmid: 20221911) |
|
[ PubMed ] [ DOI ] The National Center for Biotechnology Information (NCBI), as a primary public repository of genomic sequence data, collects and maintains enormous amounts of heterogeneous data. Data for genomes, genes, gene expressions, gene variation, gene families, proteins, and protein domains are integrated with the analytical, search, and retrieval resources through the NCBI Web site. Entrez, a text-based search and retrieval system, provides a fast and easy way to navigate across diverse biological databases.Customized genomic BLAST enables sequence similarity searches against a special collection of organism-specific sequence data and viewing the resulting alignments within a genomic context using NCBI's genome browser, Map Viewer.Comparative genome analysis tools lead to further understanding of evolutionary processes, quickening the pace of discovery. |
| Boutet et al. (2007) UniProtKB/Swiss-Prot. Methods Mol Biol 406:89-112. (pmid: 18287689) |
|
[ PubMed ] [ DOI ] The Swiss Institute of Bioinformatics (SIB), the European Bioinformatics Institute (EBI), and the Protein Information Resource (PIR) form the Universal Protein Resource (UniProt) consortium. Its main goal is to provide the scientific community with a central resource for protein sequences and functional information. The UniProt consortium maintains the UniProt KnowledgeBase (UniProtKB) and several supplementary databases including the UniProt Reference Clusters (UniRef) and the UniProt Archive (UniParc). (1) UniProtKB is a comprehensive protein sequence knowledgebase that consists of two sections: UniProtKB/Swiss-Prot, which contains manually annotated entries, and UniProtKB/TrEMBL, which contains computer-annotated entries. UniProtKB/Swiss-Prot entries contain information curated by biologists and provide users with cross-links to about 100 external databases and with access to additional information or tools. (2) The UniRef databases (UniRef100, UniRef90, and UniRef50) define clusters of protein sequences that share 100, 90, or 50% identity. (3) The UniParc database stores and maps all publicly available protein sequence data, including obsolete data excluded from UniProtKB. The UniProt databases can be accessed online (http://www.uniprot.org/) or downloaded in several formats (ftp://ftp.uniprot.org/pub). New releases are published every 2 weeks. The purpose of this chapter is to present a guided tour of a UniProtKB/Swiss-Prot entry, paying particular attention to the specificities of plant protein annotation. We will also present some of the tools and databases that are linked to each entry. |
Web servers
| Bhagwat & Aravind (2007) PSI-BLAST tutorial. Methods Mol Biol 395:177-86. (pmid: 17993673) |
|
[ PubMed ] [ DOI ] PSI-BLAST (Position-Specific Iterative Basic Local Alignment Search Tool) derives a position-specific scoring matrix (PSSM) or profile from the multiple sequence alignment of sequences detected above a given score threshold using protein-protein BLAST. This PSSM is used to further search the database for new matches, and is updated for subsequent iterations with these newly detected sequences. Thus, PSI-BLAST provides a means of detecting distant relationships between proteins. In this chapter, we discuss practical aspects of using PSI-BLAST and provide a tutorial on how to uncover distant relationships between proteins and use them to reach biologically meaningful conclusions. |
| Poupon & Janin (2010) Analysis and prediction of protein quaternary structure. Methods Mol Biol 609:349-64. (pmid: 20221929) |
|
[ PubMed ] [ DOI ] The quaternary structure (QS) of a protein is determined by measuring its molecular weight in solution. The data have to be extracted from the literature, and they may be missing even for proteins that have a crystal structure reported in the Protein Data Bank (PDB). The PDB and other databases derived from it report QS information that either was obtained from the depositors or is based on an analysis of the contacts between polypeptide chains in the crystal, and this frequently differs from the QS determined in solution.The QS of a protein can be predicted from its sequence using either homology or threading methods. However, a majority of the proteins with less than 30% sequence identity have different QSs. A model of the QS can also be derived by docking the subunits when their 3D structure is independently known, but the model is likely to be incorrect if large conformation changes take place when the oligomer assembles. |
Exercises
References
Further reading and resources
| Ulrich & Zhulin (2014) SeqDepot: streamlined database of biological sequences and precomputed features. Bioinformatics 30:295-7. (pmid: 24234005) |
|
[ PubMed ] [ DOI ] UNLABELLED: Assembling and/or producing integrated knowledge of sequence features continues to be an onerous and redundant task despite a large number of existing resources. We have developed SeqDepot-a novel database that focuses solely on two primary goals: (i) assimilating known primary sequences with predicted feature data and (ii) providing the most simple and straightforward means to procure and readily use this information. Access to >28.5 million sequences and 300 million features is provided through a well-documented and flexible RESTful interface that supports fetching specific data subsets, bulk queries, visualization and searching by MD5 digests or external database identifiers. We have also developed an HTML5/JavaScript web application exemplifying how to interact with SeqDepot and Perl/Python scripts for use with local processing pipelines. AVAILABILITY: Freely available on the web at http://seqdepot.net/. RESTaccess via http://seqdepot.net/api/v1. Database files and scripts maybe downloaded from http://seqdepot.net/download. |
| Arnold et al. (2014) SIMAP--the database of all-against-all protein sequence similarities and annotations with new interfaces and increased coverage. Nucleic Acids Res 42:D279-84. (pmid: 24165881) |
|
[ PubMed ] [ DOI ] The Similarity Matrix of Proteins (SIMAP, http://mips.gsf.de/simap/) database has been designed to massively accelerate computationally expensive protein sequence analysis tasks in bioinformatics. It provides pre-calculated sequence similarities interconnecting the entire known protein sequence universe, complemented by pre-calculated protein features and domains, similarity clusters and functional annotations. SIMAP covers all major public protein databases as well as many consistently re-annotated metagenomes from different repositories. As of September 2013, SIMAP contains >163 million proteins corresponding to ∼70 million non-redundant sequences. SIMAP uses the sensitive FASTA search heuristics, the Smith-Waterman alignment algorithm, the InterPro database of protein domain models and the BLAST2GO functional annotation algorithm. SIMAP assists biologists by facilitating the interactive exploration of the protein sequence universe. Web-Service and DAS interfaces allow connecting SIMAP with any other bioinformatic tool and resource. All-against-all protein sequence similarity matrices of project-specific protein collections are generated on request. Recent improvements allow SIMAP to cover the rapidly growing sequenced protein sequence universe. New Web-Service interfaces enhance the connectivity of SIMAP. Novel tools for interactive extraction of protein similarity networks have been added. Open access to SIMAP is provided through the web portal; the portal also contains instructions and links for software access and flat file downloads. |
| Links directory (bioinformatics.ca) [ link ] [ page ] bioinformatics.ca is the domain of the Canadian Bioinformatics Workshops, currently hosted by the Ontario Institute of Cancer research. The links directory is a curated collection of databases and services that are useful for bioinformatics and computational biology. Links are browsable in several categories, such as Model Organisms, Expression or Sequence Comparison with many subcategories. Importantly, the site contains links to all resources from the NAR database issues and the NAR web server issues in a searchable interface. The URL links to a search for the term "Systems Biology". | 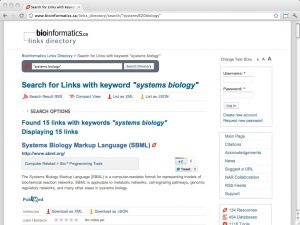 |
| Bolser et al. (2012) MetaBase--the wiki-database of biological databases. Nucleic Acids Res 40:D1250-4. (pmid: 22139927) |
|
[ PubMed ] [ DOI ] Biology is generating more data than ever. As a result, there is an ever increasing number of publicly available databases that analyse, integrate and summarize the available data, providing an invaluable resource for the biological community. As this trend continues, there is a pressing need to organize, catalogue and rate these resources, so that the information they contain can be most effectively exploited. MetaBase (MB) (http://MetaDatabase.Org) is a community-curated database containing more than 2000 commonly used biological databases. Each entry is structured using templates and can carry various user comments and annotations. Entries can be searched, listed, browsed or queried. The database was created using the same MediaWiki technology that powers Wikipedia, allowing users to contribute on many different levels. The initial release of MB was derived from the content of the 2007 Nucleic Acids Research (NAR) Database Issue. Since then, approximately 100 databases have been manually collected from the literature, and users have added information for over 240 databases. MB is synchronized annually with the static Molecular Biology Database Collection provided by NAR. To date, there have been 19 significant contributors to the project; each one is listed as an author here to highlight the community aspect of the project. |
| Dreszer et al. (2012) The UCSC Genome Browser database: extensions and updates 2011. Nucleic Acids Res 40:D918-23. (pmid: 22086951) |
|
[ PubMed ] [ DOI ] The University of California Santa Cruz Genome Browser (http://genome.ucsc.edu) offers online public access to a growing database of genomic sequence and annotations for a wide variety of organisms. The Browser is an integrated tool set for visualizing, comparing, analyzing and sharing both publicly available and user-generated genomic data sets. In the past year, the local database has been updated with four new species assemblies, and we anticipate another four will be released by the end of 2011. Further, a large number of annotation tracks have been either added, updated by contributors, or remapped to the latest human reference genome. Among these are new phenotype and disease annotations, UCSC genes, and a major dbSNP update, which required new visualization methods. Growing beyond the local database, this year we have introduced 'track data hubs', which allow the Genome Browser to provide access to remotely located sets of annotations. This feature is designed to significantly extend the number and variety of annotation tracks that are publicly available for visualization and analysis from within our site. We have also introduced several usability features including track search and a context-sensitive menu of options available with a right-click anywhere on the Browser's image. |
| Maddatu et al. (2012) Mouse Phenome Database (MPD). Nucleic Acids Res 40:D887-94. (pmid: 22102583) |
|
[ PubMed ] [ DOI ] The Mouse Phenome Project was launched a decade ago to complement mouse genome sequencing efforts by promoting new phenotyping initiatives under standardized conditions and collecting the data in a central public database, the Mouse Phenome Database (MPD; http://phenome.jax.org). MPD houses a wealth of strain characteristics data to facilitate the use of the laboratory mouse in translational research for human health and disease, helping alleviate problems involving experimentation in humans that cannot be done practically or ethically. Data sets are voluntarily contributed by researchers from a variety of institutions and settings, or in some cases, retrieved by MPD staff from public sources. MPD maintains a growing collection of standardized reference data that assists investigators in selecting mouse strains for research applications; houses treatment/control data for drug studies and other interventions; offers a standardized platform for discovering genotype-phenotype relationships; and provides tools for hypothesis testing. MPD improvements and updates since our last NAR report are presented, including the addition of new tools and features to facilitate navigation and data mining as well as the acquisition of new data (phenotypic, genotypic and gene expression). |
| NAR database issue [ link ] [ page ] Every year the journal Nucleic Acids Research (NAR) compiles a special issue on important databases in molecular biology (in January), and on important webservers and other resources (in July). The articles are peer-reviewed, and inclusion into the issue is considered a quality endorsement. Both volumes reflect the best practices in the field, as well as its rapidly changing nature. Links to databases and resources are searchable by keyword and topic in the bioinformatics.ca links directory. | 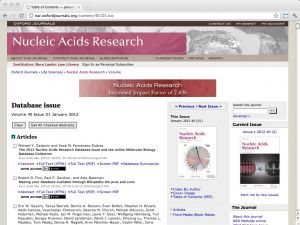 |
| NAR Web Server issue [ link ] [ page ] Every year the journal Nucleic Acids Research (NAR) compiles a special issue on important webservers in molecular biology (in July), and on important databases (in January). The articles are peer-reviewed, and inclusion into the issue is considered a quality endorsement. Both volumes reflect the best practices in the field, as well as its rapidly changing nature. Links to databases and resources are searchable by keyword and topic in the bioinformatics.ca links directory. | 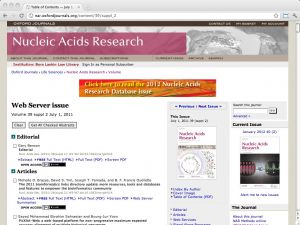 |
| The NCBI Gene database [ link ] [ page ] Gene is the NCBI's integrated database of gene information in the Entrez system. Records may include Reference Sequences (RefSeqs), maps, pathways, variations, phenotypes, compiled into the database itself, and links to genome-, phenotype-, and locus-specific resources worldwide. The URL links to the record for the human E2F1 transcription factor. For detailed information, see the Gene database information page. | 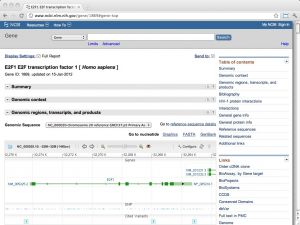 |
| Wheeler (2007) Using GenBank. Methods Mol Biol 406:23-59. (pmid: 18287687) |
|
[ PubMed ] [ DOI ] GenBank(R) is a comprehensive database of publicly available DNA sequences for more than 205,000 named organisms and for more than 60,000 within the embryophyta, obtained through submissions from individual laboratories and batch submissions from large-scale sequencing projects. Daily data exchange with the European Molecular Biology Laboratory (EMBL) in Europe and the DNA Data Bank of Japan ensures worldwide coverage. GenBank is accessible through the National Center for Biotechnology Information (NCBI) retrieval system, Entrez, which integrates data from the major DNA and protein sequence databases with taxonomy, genome, mapping, protein structure, and domain information and the biomedical journal literature through PubMed. BLAST provides sequence similarity searches of GenBank and other sequence databases. Complete bimonthly releases and daily updates of the GenBank database are available through FTP. GenBank usage scenarios ranging from local analyses of the data available through FTP to online analyses supported by the NCBI Web-based tools are discussed. To access GenBank and its related retrieval and analysis services, go to the NCBI Homepage at http://www.ncbi.nlm.nih.gov. |
| Rebhan (2010) Protein sequence databases. Methods Mol Biol 609:45-57. (pmid: 20221912) |
|
[ PubMed ] [ DOI ] Protein sequence databases do not contain just the sequence of the protein itself but also annotation that reflects our knowledge of its function and contributing residues. In this chapter, we will discuss various public protein sequence databases, with a focus on those that are generally applicable. Special attention is paid to issues related to the reliability of both sequence and annotation, as those are fundamental to many questions researchers will ask. Using both well-annotated and scarcely annotated human proteins as examples, it will be shown what information about the targets can be collected from freely available Internet resources and how this information can be used. The results are shown to be summarized in a simple graphical model of the protein's sequence architecture highlighting its structural and functional modules. |
| UniProt [ link ] [ page ] UniProt is the protein sequence database of the European Bioinformatics Institute. It is an extraordinarily well constructed, curated, and integrated resource. As a public resource, its results are freely accessible world-wide. The "Knowledge Base" (UniProtKB), which is the database proper, contains two subsections: SwissProt, the manually curated and heavily annotated protein sequence repository; it is approximately equivalent to the NCBI Refseq protein database, albeit with usually higher annotation levels. TrEMBL is much larger and contains sequences that have been computationally translated from the EMBL nucleotide sequence collection. It is approximately equivalent to the NCBI's Entrez protein database. The URL links to the entry for the Saccharomyces cerevisiae cell-cycle regulation transcription factor Mbp1. | 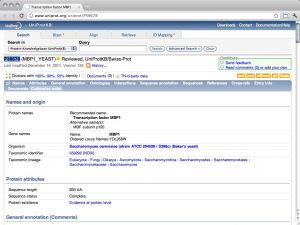 |
| Mulder (2010) Protein domain architectures. Methods Mol Biol 609:83-95. (pmid: 20221914) |
|
[ PubMed ] [ DOI ] Proteins are composed of functional units, or domains, that can be found alone or in combination with other domains. Analysis of protein domain architectures and the movement of protein domains within and across different genomes provide clues about the evolution of protein function. The classification of proteins into families and domains is provided through publicly available tools and databases that use known protein domains to predict other members in new proteins sequences. Currently at least 80% of the main protein sequence databases can be classified using these tools, thus providing a large data set to work from for analyzing protein domain architectures. Each of the protein domain databases provide intuitive web interfaces for viewing and analyzing their domain classifications and provide their data freely for downloading. Some of the main protein family and domain databases are described here, along with their Web-based tools for analyzing domain architectures. |
| Laskowski (2011) Protein structure databases. Mol Biotechnol 48:183-98. (pmid: 21225378) |
|
[ PubMed ] [ DOI ] Web-based protein structure databases come in a wide variety of types and levels of information content. Those having the most general interest are the various atlases that describe each experimentally determined protein structure and provide useful links, analyses and schematic diagrams relating to its 3D structure and biological function. Also of great interest are the databases that classify 3D structures by their folds as these can reveal evolutionary relationships which may be hard to detect from sequence comparison alone. Related to these are the numerous servers that compare folds-particularly useful for newly solved structures, and especially those of unknown function. Beyond these there are a vast number of databases for the most specialized user, dealing with specific families, diseases, structural features and so on. |
| SGD: Saccharomyces Genome Database [ link ] [ page ] The Saccharomyces genome database is a curated database that integrates sequence, structure and function information for yeast molecular biology. It is one of the important model organism databases and can be considered a paradigm for the entire field. The url links to the information page of the cell-cycle regulation transcription factor Mbp1. | 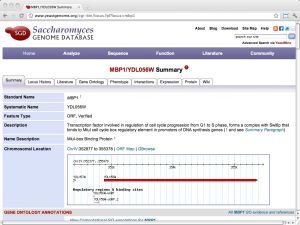 |
| MGI (Mouse Genome Informatics) [ link ] [ page ] The model organism database MGI (Mouse Genome Informatics) is the primary community database resource for the laboratory mouse. It integrates genomics, expression, tumor biology and metabolism information and actively curates GO annotations for mouse genes. The stated goal is to enhance the utility of mouse research for the study of human health and disease. For example, wherever available, human orthologues are cross-referenced with the respective mouse genes. The URL links to the gene details of the mouse orthologue of human E2F1. | 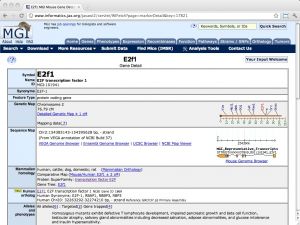 |
|
GO: the Gene Ontology project [ link ] [ page ] Ontologies are important tools to organize and compute with non-standardized information, such as gene annotations. The Gene Ontology project (GO) constructs ontologies for gene and gene product attributes across numerous species. Three major ontologies are being developed: molecular process, biological function and cellular location. Each includes terms, their definition, and their relationships. In addition, genes and gene products are being been annotated with their GO terms and the type of evidence that underlies the annotation. A number of tools such as the AmiGO browser are available to analyse relationships, construct ontologies and curate annotations. Data can be freely downloaded in formats that are convenient for computation. |  |
| The Gene Wiki project [ link ] [ page ] The Gene Wiki project aims to create Wikipedia articles for every human gene whose function has been assigned. This provides pages that are ideally suited for free, community-driven, integrated information resources. Access to the project is through the Gene Wiki Portal, which contains guidelines for contributors. The pages are easy to find since they are linked to the HGNC recognized gene name. For example, the URL links to the human E2F1 transcription factor page. |  |
| Gene/Protein Synonym Database [ link ] [ page ] The ExPASy hosted Gene/Protein Synonym Database collects gene name synonyms from the majority of model organism databases and UniProt, cross-references them and provides a searchable interface. |  |
| HUGO Gene Nomenclature Committee [ link ] [ page ] The HUGO Gene Nomenclature Committee (HGNC) has assigned unique gene symbols and names to more than 32,000 human loci, of which over 19,000 are protein coding. genenames.org is a curated online repository of HGNC-approved gene nomenclature and associated resources including links to genomic, proteomic and phenotypic information, as well as dedicated gene family pages. This site is the definitive resource to resolve gene name ambiguities. The URL links to the search results for Rbp3, which is both a deprecated synonym for the human E2F transcription factor 1, and the official name of retinol binding protein 3. |  |
| Ooi et al. (2010) Databases of protein-protein interactions and complexes. Methods Mol Biol 609:145-59. (pmid: 20221918) |
|
[ PubMed ] [ DOI ] In the current understanding, translation of genomic sequences into proteins is the most important path for realization of genome information. In exercising their intended function, proteins work together through various forms of direct (physical) or indirect interaction mechanisms. For a variety of basic functions, many proteins form a large complex representing a molecular machine or a macromolecular super-structural building block. After several high-throughput techniques for detection of protein-protein interactions had matured, protein interaction data became available in a large scale and curated databases for protein-protein interactions (PPIs) are a new necessity for efficient research. Here, their scope, annotation quality, and retrieval tools are reviewed. In addition, attention is paid to portals that provide unified access to a variety of such databases with added annotation value. |
| Ooi et al. (2010) Biomolecular pathway databases. Methods Mol Biol 609:129-44. (pmid: 20221917) |
|
[ PubMed ] [ DOI ] From the database point of view, biomolecular pathways are sets of proteins and other biomacromolecules that represent spatio-temporally organized cascades of interactions with the involvement of low-molecular compounds and are responsible for achieving specific phenotypic biological outcomes. A pathway is usually associated with certain subcellular compartments. In this chapter, we analyze the major public biomolecular pathway databases. Special attention is paid to database scope, completeness, issues of annotation reliability, and pathway classification. In addition, systems for information retrieval, tools for mapping user-defined gene sets onto the information in pathway databases, and their typical research applications are reviewed. Whereas today, pathway databases contain almost exclusively qualitative information, the desired trend is toward quantitative description of interactions and reactions in pathways, which will gradually enable predictive modeling and transform the pathway databases into analytical workbenches. |
| Schomburg & Schomburg (2010) Enzyme databases. Methods Mol Biol 609:113-28. (pmid: 20221916) |
|
[ PubMed ] [ DOI ] Enzymes are catalysts for the chemical reactions in the metabolism of all organisms and play a key role in the regulation of metabolic steps within the cells, as drug targets, and in a wide range of biotechnological applications. With respect to reaction type, they are grouped into six classes, namely oxidoreductases, transferases, hydrolases, lyases, and ligases. EC-Numbers are assigned by the IUBMB. Enzyme functional databases cover a wide range of properties and functions, such as occurrence, kinetics of enzyme-catalyzed reactions, structure, or metabolic function. BRENDA stores a large variety of different data for all classified enzymes whereas KEGG, MEROPS, MetaCyc, REBASE, CAzy, ESTHER, PeroxiBase, and KinBase specialize in either certain aspects of enzyme function or specific enzyme classes, organisms, or metabolic pathways. Databases covering enzyme nomenclature are ExplorEnz, SIB-ENZYME, and IntEnz. |
| Reactome [ link ] [ page ] Reactome is a multi-site collaboration to develop an open source, curated bioinformatics database of human pathways and reactions. It includes annotations, pathways and tools for pathway browsing and analysis, including pathway assignment and overrepresentation analysis of user-supplied data sets. Making use of orthology prediction, Reactome also provides cross-species pathway inference for a large number of model organisms. The URL accesses the E2F mediated regulation of DNA replication. | 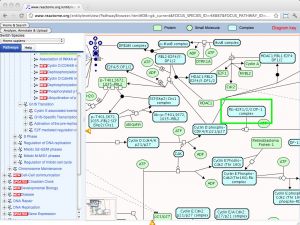 |
| KEGG [ link ] [ page ] The Kyoto Encyclopedia of Genes and Genomes (KEGG) is a deeply curated resource that integrates genomic, chemical and systemic functional information. Regrettably, ftp access to this resource is no longer free and personal-use academic licenses outside of Japan are required to purchase a license at the cost of USD 2,000 per year (as per January 2012) from a non-profit organization, founded to ensure the long-term survival of the database. (Read here about the background that explains how public funding is falling short of sustainable levels, thus jeopardizing the ongoing curation and software development activities - and thereby the entire investment into the resource). As of now, use of the Web resources is not to affected. KEGG contains several sections of systems-, genome- and small molecule related information. See here for an overview. The URL links to the pathway map of the yeast cell-cycle. | 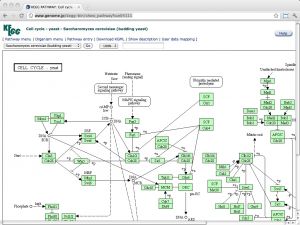 |
| BioCyc [ link ] [ page ] BioCyc is a collection of metabolic pathways databases, derived from computational annotation (and manual curation) of whole-genome sequence data. The database range from highly curated (such as EcoCyc, and HumanCyc, and the comparative, multiorganism MetaCyc resource) to purely computationally derived. Searches can be performed by component, reaction or pathway, and by ontology. The example URL leads to the cellulosome cellulose degradation pathway in MetaCyc. | 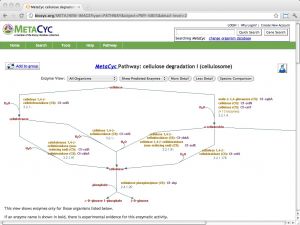 |
| GMOD Generic Model Organism Database project [ link ] [ page ] GMOD (the Generic Model Organism Database project), is a collection of open source software tools for creating and managing genome-scale biological databases. GMOD tools are in use at many large and small community databases, especially for Model Organisms. The include the genome browser GBrowse, the CHADO relational database, the GFF annotation databases, and much more The goal is to free developers of community scale biomolecualr databases from reinventing the wheel. A good overview of resources and principles is available on the GMOD wiki. |  |