Lecture 07
Jump to navigation
Jump to search































(Previous lecture) ... (Next lecture)
Multiple Sequence Alignment
Objectives for this part of the course
- Understand that MSA is an unsolved, difficult problem with different "best" solutions for different purposes.
- Be familiar with different biological heuristics that distinguish a "good" alignment from a "poor" alignment.
- Understand the importance of benchmarks for assessing the performance of computational tools.
- Be aware of how different biological priorities have resulted in different algorithmic strategies and some of the available tools that represent them.
- Be aware that the most frequently used and referenced tool - CLUSTAL - is no longer state-of-the-art and know which modern tools are much better.
- Confidently be able to survey recent developments and choose an appropriate algorithm.
- Be able to perform and interpret MSAs in practice, know how to prepare input, which formats to use and what common output formats look like.
- Understand strategies to prepare input and improve alignments, based on the requirement of columnwise homology.
- Know about strategies and tools for manual editing of alignments.
Links summary
- Dallas PROMALS Web server
- EBI CLUSTAL web server
- EBI T-Coffee Web server
- EBI MUSCLE Web server
- Stanford PROBCONS server
- Indiana SPEM server
- MUSCA, based on the Teiresias pattern discovery algorithm
- HMMER, a profile hidden Markov model tool
- BAliBASE, BAliBASE 2.0 and BAliBASE 3.0
- EBI help page on formats
- Jalview home page
- Embnet BOXSHADE server
- Wikipedia page on Multiple Sequence Alignment
Exercises
- Read Cedric Notredame's MSA review (2007)
- Read Edgar and Batzoglou's MSA review (2005)
- More exercises will be covered in Assignment 3.
Lecture slides
Uses and Problems
Slide 004

Lecture 07, Slide 004
Multiple sequence alignments don't only match residues. They also give information on how strongly a residue is conserved, what it can be replaced with, which species share particular sequence patterns, and where in the sequence indels can be tolerated. An analysis of conservation even allows to distinguish between structurally and functionally conserved residues!
Multiple sequence alignments don't only match residues. They also give information on how strongly a residue is conserved, what it can be replaced with, which species share particular sequence patterns, and where in the sequence indels can be tolerated. An analysis of conservation even allows to distinguish between structurally and functionally conserved residues!
Slide 005

Lecture 07, Slide 005
Multiple sequence alignments are more accurate than pairwise alignments, thus they are the method of choice for starting homology modeling projects. Their combined information is invaluable for secondary structure prediction and sensitive database searches. They contain the information needed for inferences about evolutionary relationships, i.e. the order in which particular sequence changes occurred.
Multiple sequence alignments are more accurate than pairwise alignments, thus they are the method of choice for starting homology modeling projects. Their combined information is invaluable for secondary structure prediction and sensitive database searches. They contain the information needed for inferences about evolutionary relationships, i.e. the order in which particular sequence changes occurred.
Slide 006

Lecture 07, Slide 006
Multiple alignments cannot necessarily be constructed from pairwise alignments. Moreover, it may be impossible to merge three mutually pairwise alignments into a non-contradicting multiple alignment. However the inverse is always possible: a multiple alignment can be decomposed into pairwise alignments.
Multiple alignments cannot necessarily be constructed from pairwise alignments. Moreover, it may be impossible to merge three mutually pairwise alignments into a non-contradicting multiple alignment. However the inverse is always possible: a multiple alignment can be decomposed into pairwise alignments.
Slide 007

Lecture 07, Slide 007
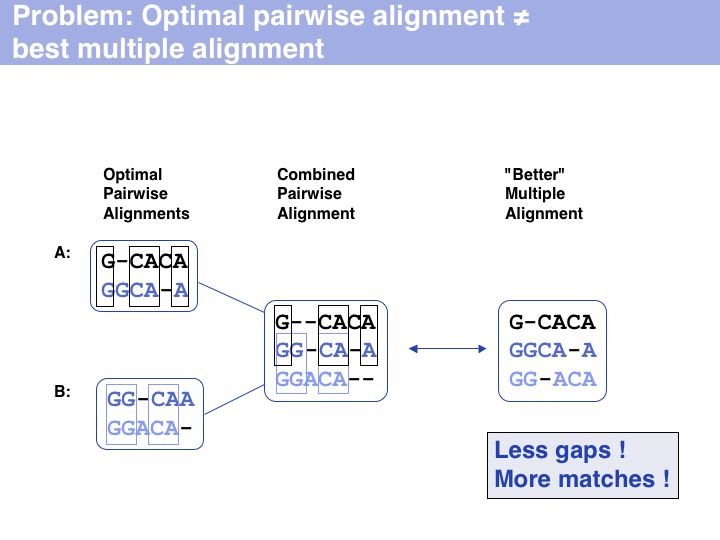
Besides being intractable, it is questionable how meaningful the objective function of optimal sequence alignments - the pair score - is for multiple alignments. For example the pair score does not refelct on the pattern of indel placements, or whether a particular motif is well-conserved.
Besides being intractable, it is questionable how meaningful the objective function of optimal sequence alignments - the pair score - is for multiple alignments. For example the pair score does not refelct on the pattern of indel placements, or whether a particular motif is well-conserved.
Right, wrong, good and poor
Slide 009

Lecture 07, Slide 009
If we want to write an algorithm to optimize anything at all, we first must define how we can measure the quality of the result. This metric defines the target function or objective function.
If we want to write an algorithm to optimize anything at all, we first must define how we can measure the quality of the result. This metric defines the target function or objective function.
Slide 010

Lecture 07, Slide 010
In practice, each of the biological objectives we can define suggests a different alignment strategy. The most modern algorithms currently available attempt to satisfy these heuristics simultaneously. Note that these are heuristics, they are not the result of some rigorously applied theory, but reflect the complex relationship between protein sequence, structure, evolution and selection.
In practice, each of the biological objectives we can define suggests a different alignment strategy. The most modern algorithms currently available attempt to satisfy these heuristics simultaneously. Note that these are heuristics, they are not the result of some rigorously applied theory, but reflect the complex relationship between protein sequence, structure, evolution and selection.
Slide 011

Lecture 07, Slide 011
MSA in practice
Slide 013

Lecture 07, Slide 013
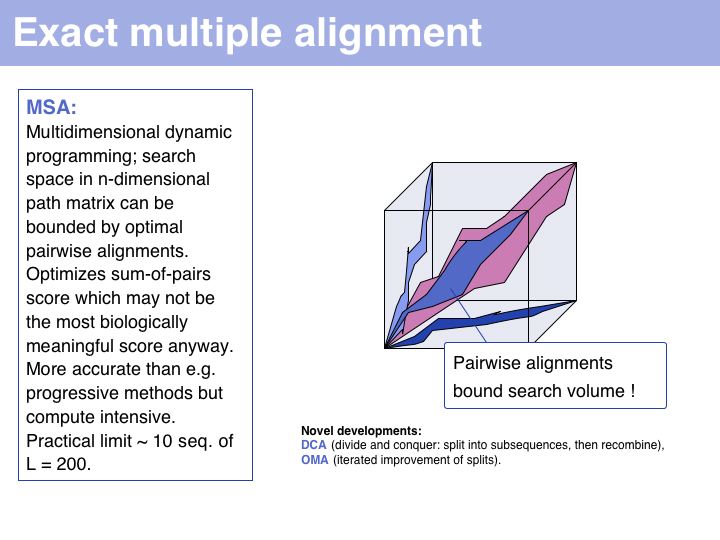
Exact methods certainly have their place where it comes to analyzing and improving algorithms; they are especially of interest to computer science because high-dimensional optimal alignment is a difficult problem. However they cannot compete in terms of result-quality with modern heuristic methods.
Exact methods certainly have their place where it comes to analyzing and improving algorithms; they are especially of interest to computer science because high-dimensional optimal alignment is a difficult problem. However they cannot compete in terms of result-quality with modern heuristic methods.
Slide 014

Lecture 07, Slide 014
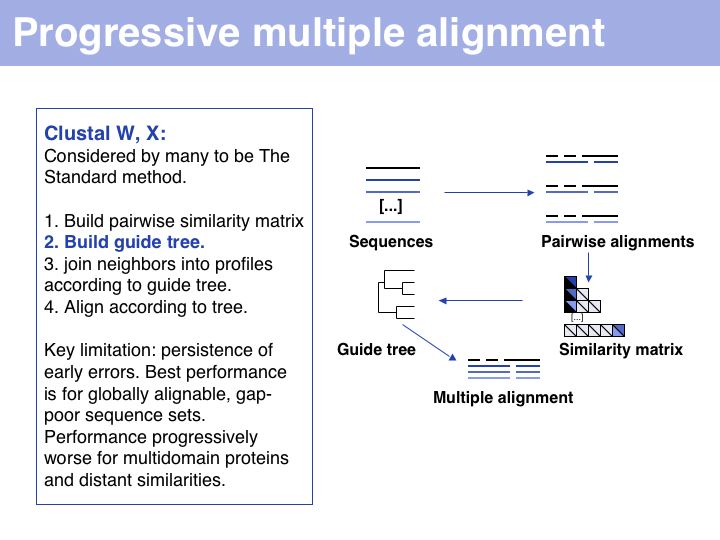
Progressive alignment is one of three fundamental algorithmic approaches to MSA. The EBI offers Clustal alignments online.
Progressive alignment is one of three fundamental algorithmic approaches to MSA. The EBI offers Clustal alignments online.
Slide 015

Lecture 07, Slide 015
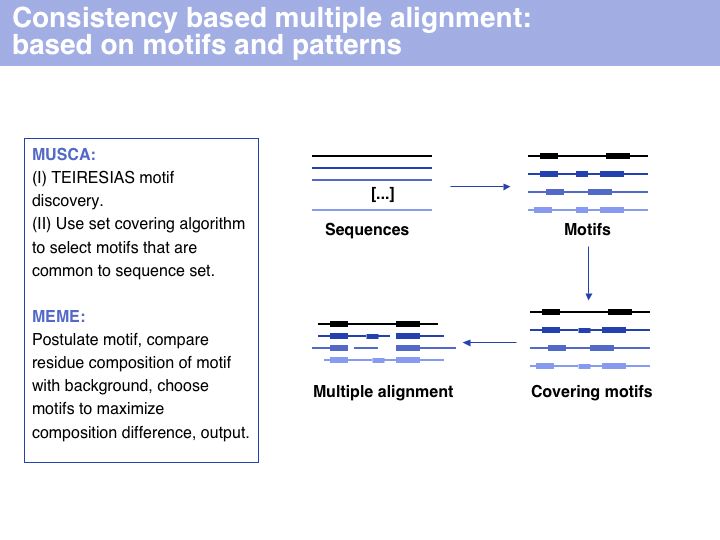
Consistency based multiple alignment is one of three fundamental algorithmic approaches to MSA. Many modern algorithms have a consistency based step included, however none of them relies solely on consistency, since problems from spurious local similarity can corrupt the alignment. MUSCA, based on the Teiresias pattern discovery algorithm is offered through IBM's Watson Labs Web server. Similarly, the MEME algorithm for motif discoverywhich is more commonly used in sequence analysis infers a motif-based alignment.
Consistency based multiple alignment is one of three fundamental algorithmic approaches to MSA. Many modern algorithms have a consistency based step included, however none of them relies solely on consistency, since problems from spurious local similarity can corrupt the alignment. MUSCA, based on the Teiresias pattern discovery algorithm is offered through IBM's Watson Labs Web server. Similarly, the MEME algorithm for motif discoverywhich is more commonly used in sequence analysis infers a motif-based alignment.
Slide 016

Lecture 07, Slide 016
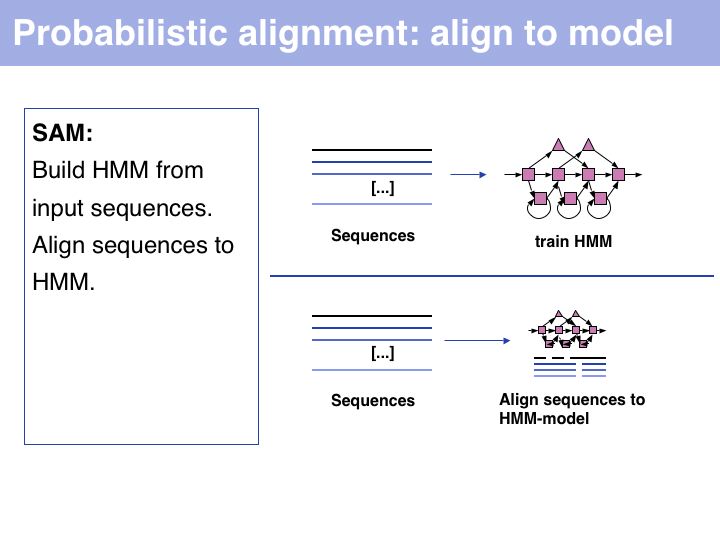
Probabilistic' multiple alignment is one of three fundamental algorithmic approaches to MSA. A statistical model of the sequences is built, then the alignment can be generated by aligning the sequences to the model. Of course, aligning sequences to a profile is a special case of this procedure: PSI BLAST can thus be used as an alignment algorithm. The most widely used algortihm is Sean Eddy's HMMER, a profile hidden Markov model tool which is also used in the generation of the Pfam domain database.
Probabilistic' multiple alignment is one of three fundamental algorithmic approaches to MSA. A statistical model of the sequences is built, then the alignment can be generated by aligning the sequences to the model. Of course, aligning sequences to a profile is a special case of this procedure: PSI BLAST can thus be used as an alignment algorithm. The most widely used algortihm is Sean Eddy's HMMER, a profile hidden Markov model tool which is also used in the generation of the Pfam domain database.
Slide 017

Lecture 07, Slide 017
Altschul et al. (1998) Nucleic Acids Research 25:3389-3402
Altschul et al. (1998) Nucleic Acids Research 25:3389-3402
Slide 019

Lecture 07, Slide 019
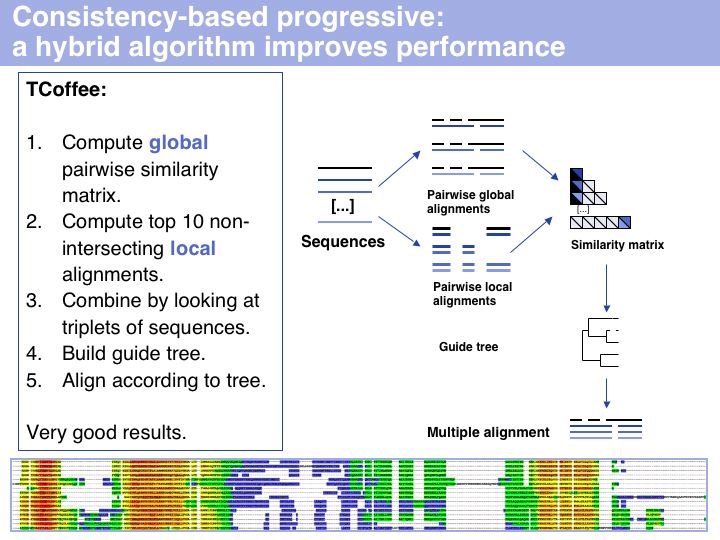
I personally rate TCoffee as the most useful and useable tool that is currently available. It is robust, fast, and gives reasonable results for many cases. Usually it is very noticeably better then CLUSTAL and I would reject any result based on CLUSTAL for that reason. The EBI offers a T-Coffee Web server which is very easy to use (although alignment size is limited;). Source code can be obtained and a local installation on UNIX machines is straightforward. The TCoffee Web page links to another Web server and also offers 3DCoffee, a variant that automatically fetches related structures and incorporates structural alignments for increased accuracy.
The inset image shows one of the usuful features of TCoffee: an alignment output in which sequence is coloured according to the local quality of the alignment. This makes reliable and unreliable regions easy to spot, and immediately highlights outliers that could for example be due to sequence errors, such as frameshifts in exons. (MSA taken from the Mbp1 full-length alignment).
I personally rate TCoffee as the most useful and useable tool that is currently available. It is robust, fast, and gives reasonable results for many cases. Usually it is very noticeably better then CLUSTAL and I would reject any result based on CLUSTAL for that reason. The EBI offers a T-Coffee Web server which is very easy to use (although alignment size is limited;). Source code can be obtained and a local installation on UNIX machines is straightforward. The TCoffee Web page links to another Web server and also offers 3DCoffee, a variant that automatically fetches related structures and incorporates structural alignments for increased accuracy.
The inset image shows one of the usuful features of TCoffee: an alignment output in which sequence is coloured according to the local quality of the alignment. This makes reliable and unreliable regions easy to spot, and immediately highlights outliers that could for example be due to sequence errors, such as frameshifts in exons. (MSA taken from the Mbp1 full-length alignment).
Slide 020

Lecture 07, Slide 020
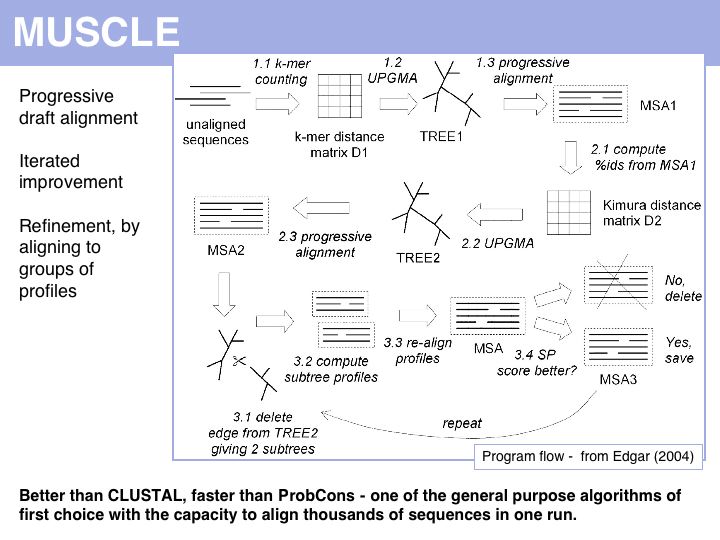
Run the MUSCLE MSAs via the EBI MUSCLE Web server which is very easy to use, or via the Berkeley MUSCLE server courtesy of Kimmen Sjolander's lab. Source code and compiled code can be obtained from the Muscle homepage and a local installation on UNIX and Windows machines is straightforward. The site also hosts the PREFAB multiple alignment benchmark.
Run the MUSCLE MSAs via the EBI MUSCLE Web server which is very easy to use, or via the Berkeley MUSCLE server courtesy of Kimmen Sjolander's lab. Source code and compiled code can be obtained from the Muscle homepage and a local installation on UNIX and Windows machines is straightforward. The site also hosts the PREFAB multiple alignment benchmark.
Slide 021

Lecture 07, Slide 021
One of the best algorithms that aligns sequences without additional database information. Run it on the web via the Stanford PROBCONS server, or download the code and install locally.
One of the best algorithms that aligns sequences without additional database information. Run it on the web via the Stanford PROBCONS server, or download the code and install locally.
Slide 022

Lecture 07, Slide 022
SPEM is one of the most accurate algorithms currently available, in particular for sequences of very low similarity. Run alignments via the Indiana SPEM server.
SPEM is one of the most accurate algorithms currently available, in particular for sequences of very low similarity. Run alignments via the Indiana SPEM server.
Slide 023

Lecture 07, Slide 023
One of the latest additions to the toolkit, PROMALS is currently the most accurate MSA tool available. Run it on the Dallas PROMALS Web server. Read the PROMALS paper in the 2007 NAR Web server issue.
One of the latest additions to the toolkit, PROMALS is currently the most accurate MSA tool available. Run it on the Dallas PROMALS Web server. Read the PROMALS paper in the 2007 NAR Web server issue.
Slide 024

Lecture 07, Slide 024
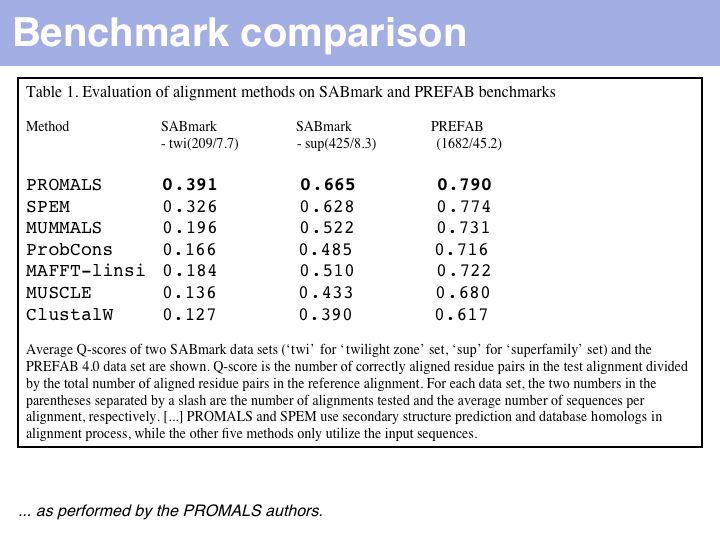
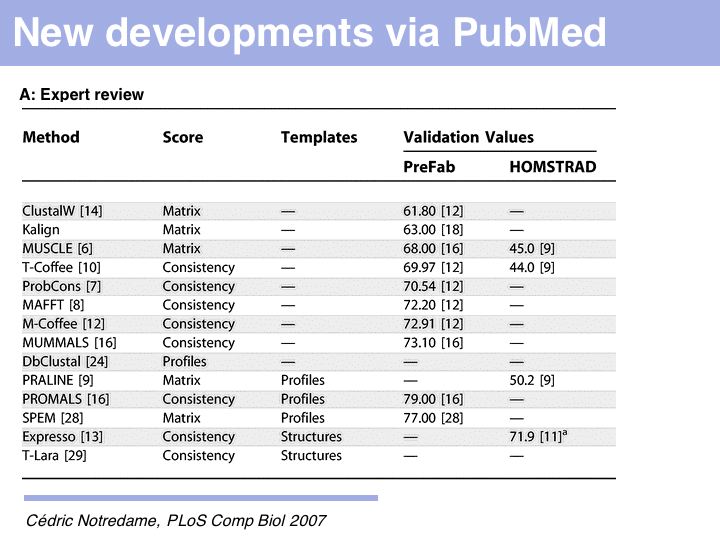
Just what does PROMALS' improved performance mean, relative to e.g. CLUSTAL? For one, we can see a clear leap in performance through the inclusion of database information and consensus structure predictions (SPEM and PROMALS). On the other hand, regarding the SABmark superfamily dataset that is perhaps most characteristic of "typical" alignment problems with recognizeable, but low % identity, PROMALS achieves a 50% improvement relative to CLUSTAL, a 30% improvement relative to MUSCLE and ProbCons. This is much more than just statistical noise.
Just what does PROMALS' improved performance mean, relative to e.g. CLUSTAL? For one, we can see a clear leap in performance through the inclusion of database information and consensus structure predictions (SPEM and PROMALS). On the other hand, regarding the SABmark superfamily dataset that is perhaps most characteristic of "typical" alignment problems with recognizeable, but low % identity, PROMALS achieves a 50% improvement relative to CLUSTAL, a 30% improvement relative to MUSCLE and ProbCons. This is much more than just statistical noise.
Slide 025

Lecture 07, Slide 025
How do we know that a new algorithm is better than a previous one? Benchmarks, or "Gold Standards" are an essential part of scientific hygiene. We as users must demand objective comparisons to existing methods, as referees we must require them for publication, as members of the research community we must participate in defining them and provide raw data for their construction. But we must also realize that an "arms-race" of sorts may be ensuing: as developers use the benchmarks as a training set, artificially high performance scores may be generated and performance on novel problems may degrade.
How do we know that a new algorithm is better than a previous one? Benchmarks, or "Gold Standards" are an essential part of scientific hygiene. We as users must demand objective comparisons to existing methods, as referees we must require them for publication, as members of the research community we must participate in defining them and provide raw data for their construction. But we must also realize that an "arms-race" of sorts may be ensuing: as developers use the benchmarks as a training set, artificially high performance scores may be generated and performance on novel problems may degrade.
Slide 026

Lecture 07, Slide 026
Access the original BAliBASE (1999) here. Two updated versions have been created: BAliBASE 2.0 (2000) and BAliBASE 3.0. Central to BAliBASE is the concept of core blocks of alignable regions in which a pairwise correspondence of residues can be defined; outside these regions an alignment is not possible since the structural differences are too large. (BAliBASE: Thompson J. et al., (1999) Bioinformatics 15:87-88.).
Access the original BAliBASE (1999) here. Two updated versions have been created: BAliBASE 2.0 (2000) and BAliBASE 3.0. Central to BAliBASE is the concept of core blocks of alignable regions in which a pairwise correspondence of residues can be defined; outside these regions an alignment is not possible since the structural differences are too large. (BAliBASE: Thompson J. et al., (1999) Bioinformatics 15:87-88.).
Slide 027

Lecture 07, Slide 027
SABmark: Van Walle et al. (2005) Bioinformatics 21:1267-1268. SABmark homepage.
SABmark: Van Walle et al. (2005) Bioinformatics 21:1267-1268. SABmark homepage.
Slide 028

Lecture 07, Slide 028
Construction of PREFAB is described in MUSCLE: Edgar (2004) Nucl Acids Res 32:1792-1797.
Construction of PREFAB is described in MUSCLE: Edgar (2004) Nucl Acids Res 32:1792-1797.
Slide 029

Lecture 07, Slide 029
Slide 030

Lecture 07, Slide 030
Slide 031

Lecture 07, Slide 031
"Relevance" for Google may not be the same as relevance for your work. For some applications, novelty is more important than cross-references and page-hits. For a more curated view, you can try the Wikipedia page on Multiple Sequence Alignment or the Wikiomics page. (Wikiomics is a project you should know about, but it doesn't appear to be catching on very well.)
"Relevance" for Google may not be the same as relevance for your work. For some applications, novelty is more important than cross-references and page-hits. For a more curated view, you can try the Wikipedia page on Multiple Sequence Alignment or the Wikiomics page. (Wikiomics is a project you should know about, but it doesn't appear to be catching on very well.)
Slide 032

Lecture 07, Slide 032
The obvious first approach is to search for a recent review. For the last year of sequence alignment literature in PubMed: search ("multiple sequence alignment"[ti] OR "multiple alignment"[ti]) AND (server OR algorithm) AND "last 1 years"[dp] or just click here. Note that not all "reviews" have been tagged by the PubMed curators as such. In the list returned in September 2007, the most recent review was found by the above search strategy, but it was in the list of publications, not in the sub-set of reviews. Of course, no recent review may be available, or the available reviews may not be very informative. Cedric Notredame's MSA review (2007) is technical and probably less-helpful for the non-expert, although it emphasizes the paradigm shift towards template based alignment strategies well. Edgar and Batzoglou's MSA review (2005), by the authors of MUSCLE and ProbCons, is much more readable and a good, comprehensive introduction to modern methods.
The obvious first approach is to search for a recent review. For the last year of sequence alignment literature in PubMed: search ("multiple sequence alignment"[ti] OR "multiple alignment"[ti]) AND (server OR algorithm) AND "last 1 years"[dp] or just click here. Note that not all "reviews" have been tagged by the PubMed curators as such. In the list returned in September 2007, the most recent review was found by the above search strategy, but it was in the list of publications, not in the sub-set of reviews. Of course, no recent review may be available, or the available reviews may not be very informative. Cedric Notredame's MSA review (2007) is technical and probably less-helpful for the non-expert, although it emphasizes the paradigm shift towards template based alignment strategies well. Edgar and Batzoglou's MSA review (2005), by the authors of MUSCLE and ProbCons, is much more readable and a good, comprehensive introduction to modern methods.
Slide 033

Lecture 07, Slide 033
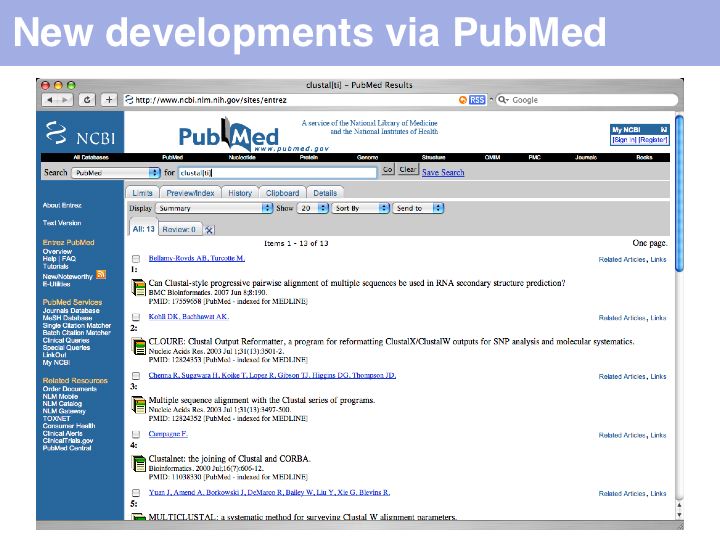
An alternative and more exploratory approach is to choose a recent highly relevant article, then to use the NCBI's "Related Articles" service. This search strategy allows you to search forward in time from a particular publication. In the above example, a serch for clustal[ti] yielded a publication on CLUSTAL from 2003 ...
An alternative and more exploratory approach is to choose a recent highly relevant article, then to use the NCBI's "Related Articles" service. This search strategy allows you to search forward in time from a particular publication. In the above example, a serch for clustal[ti] yielded a publication on CLUSTAL from 2003 ...
Slide 034

Lecture 07, Slide 034
... in the list of related articles (in September 2007) the article on PROMALS (2007): was number 5 in the hit-list, SPEM (2005) came as number 50.
... in the list of related articles (in September 2007) the article on PROMALS (2007): was number 5 in the hit-list, SPEM (2005) came as number 50.
Slide 035

Lecture 07, Slide 035
Spend some time and thought before you run the MSA to review the sequences that you are planning to align. Including un-alignable sequence will lead the algorithms astray and has the potential to degrade the entire alignment.
Spend some time and thought before you run the MSA to review the sequences that you are planning to align. Including un-alignable sequence will lead the algorithms astray and has the potential to degrade the entire alignment.
Editing and printing
Slide 037

Lecture 07, Slide 037
Three common formats exist for MSA results. An aligned multi FASTA file contains FASTA formatted sequences into which gap characters have been inserted. Of course, multi FASTA files can also be unaligned and they are the most common way of formatting input files for MSAs.
Three common formats exist for MSA results. An aligned multi FASTA file contains FASTA formatted sequences into which gap characters have been inserted. Of course, multi FASTA files can also be unaligned and they are the most common way of formatting input files for MSAs.
Slide 038

Lecture 07, Slide 038
Three common formats exist for MSA results. MSF is a legacy format from the GCG package of sequence alignments, also produced by the EMBOSS tool EMMA, and supported as a valid input format for many programs. Gaps are denoted by periods and checksums are calculated for the sequences and for the alignment.
Three common formats exist for MSA results. MSF is a legacy format from the GCG package of sequence alignments, also produced by the EMBOSS tool EMMA, and supported as a valid input format for many programs. Gaps are denoted by periods and checksums are calculated for the sequences and for the alignment.
Slide 039

Lecture 07, Slide 039
Three common formats exist for MSA results. A CLUSTAL formatted alignment is the format in most common use. Take care when formatting input files to ensure the first 10 characters in your input file are unique and contain no special characters! I have seen programs break on blanks, hyphens and | (pipe). The latter is especially annoying, since the | character is used in NCBI FASTA files to separate the database identifier from the accession number. (More information at the EBI help page on formats.)
Three common formats exist for MSA results. A CLUSTAL formatted alignment is the format in most common use. Take care when formatting input files to ensure the first 10 characters in your input file are unique and contain no special characters! I have seen programs break on blanks, hyphens and | (pipe). The latter is especially annoying, since the | character is used in NCBI FASTA files to separate the database identifier from the accession number. (More information at the EBI help page on formats.)
Slide 040

Lecture 07, Slide 040
It is common and perfectly permissible to manually edit a MSA with some biologically motivated heuristic in mind as long as you document what you have done! In the early days of MSAs, editing was simply required since the results were often obviously inadequate. In all cases in which the algorithm uses only the input sequences for the alignment, this still holds true. However, regarding the more modern template-based procedures (e.g. SPEM, PROMALS or PRALINE) I would be more reluctant to edit, since we may be actively ignoring/discarding the additional information the algorithm has used.
It is common and perfectly permissible to manually edit a MSA with some biologically motivated heuristic in mind as long as you document what you have done! In the early days of MSAs, editing was simply required since the results were often obviously inadequate. In all cases in which the algorithm uses only the input sequences for the alignment, this still holds true. However, regarding the more modern template-based procedures (e.g. SPEM, PROMALS or PRALINE) I would be more reluctant to edit, since we may be actively ignoring/discarding the additional information the algorithm has used.
Slide 041

Lecture 07, Slide 041
Slide 042

Lecture 07, Slide 042
Jalview is integrated into the EBI multiple sequence alignment services, or you can access Jalview home page.
Jalview is integrated into the EBI multiple sequence alignment services, or you can access Jalview home page.
Slide 043

Lecture 07, Slide 043
VMD structural alignment: Eargle et al. (2006) Bioinformatics 22:504-506
VMD structural alignment: Eargle et al. (2006) Bioinformatics 22:504-506
Slide 044

Lecture 07, Slide 044
The PFAAT homepage
The PFAAT homepage
Slide 045

Lecture 07, Slide 045
The CINEMA homepage.
The CINEMA homepage.
Slide 046

Lecture 07, Slide 046
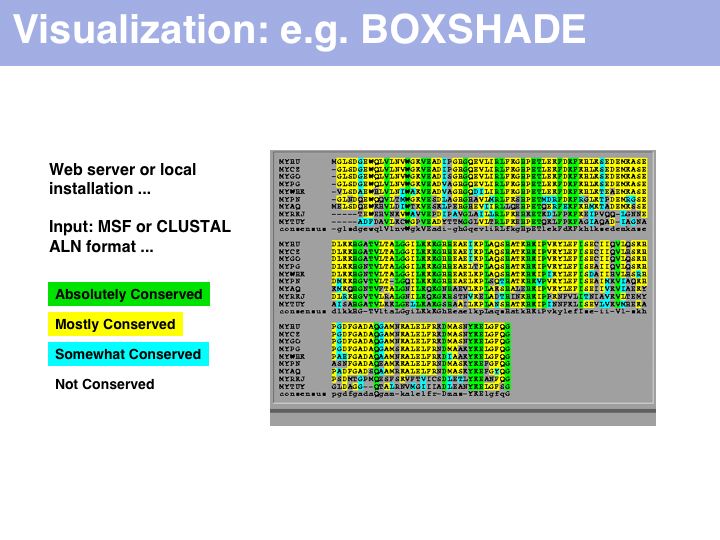
Purely for alignment visualization, run it from the Embnet BOXSHADE server or the Pasteur institute BOXSHADE server. The EMBOSS package has tools with similar functionality.
Purely for alignment visualization, run it from the Embnet BOXSHADE server or the Pasteur institute BOXSHADE server. The EMBOSS package has tools with similar functionality.
Slide 047

Lecture 07, Slide 047
Slide 048

Lecture 07, Slide 048
Slide 049

Lecture 07, Slide 049
Slide 050

Lecture 07, Slide 050