Difference between revisions of "Lecture 08"
Jump to navigation
Jump to search

























| (2 intermediate revisions by the same user not shown) | |||
| Line 1: | Line 1: | ||
| + | <!-- div style="padding: 5px; background: #FF4560; border:solid 2px #000000;"> | ||
| + | '''Update Warning!''' | ||
| + | This page has not been revised yet for the 2008 Fall term. | ||
| + | Some of the slides will probably be reused, but please consider the page as a whole out of date | ||
| + | as long as this warning appears here. Also, the lectures may be taught in a different sequence. | ||
| + | </div --> | ||
| + | | ||
| + | | ||
__NOTOC__ | __NOTOC__ | ||
| − | <small>[[Lecture_07|(Previous lecture)]] ... [[Lecture_09|(Next lecture)]]</small> | + | <small>[[Lecture_07|(Previous lecture)]] ... [[Lecture_09|(Next lecture)]]</small> |
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | == | + | <br> |
| + | <br> | ||
| + | <div style="padding: 2px; background: #879BFA; border:solid 1px #AAAAAA;"> | ||
| + | ==Fast Sequence Database Searches== | ||
| + | </div><br> | ||
| + | | ||
| − | |||
| − | |||
| − | + | <br> | |
| − | ====== | + | <div style="padding: 10 px; background: #B0B8D7; border:solid 1px #AAAAAA;"> |
| − | [[ | + | ====Objectives for this part of the course==== |
| + | </div><br> | ||
| + | * Understand the advantages and limitations of heuristic, local alignment vs. optimal alignment.<br> | ||
| + | * Initiate a BLAST search.<br> | ||
| + | * Understand different BLAST algorithms and for which computational task they are appropriate.<br> | ||
| + | * Understand the contents of the different databases offered by the NCBI for BLASTing and be able to restrict a search by database and organism.<br> | ||
| + | * Understand how to set the algorithm's parameters for different purposes.<br> | ||
| + | * Understand all information in a BLAST report.<br> | ||
| + | * Be able to evaluate the significance of hits through E-values and other metrics / features of the alignment.<br> | ||
| + | * Be able to use PSI-BLAST and avoid and recognize profile corruption; be able to evaluate E-value trends of questionable alignments.<br> | ||
| + | * Be familiar with novel developments beyond BLAST. | ||
| + | |||
| + | |||
| + | <br> | ||
| + | <div style="padding: 10 px; background: #B0B8D7; border:solid 1px #AAAAAA;"> | ||
| + | ====Links summary==== | ||
| + | </div><br> | ||
| + | *[http://www.ncbi.nlm.nih.gov/BLAST/ '''BLAST''']<br> | ||
| + | *[http://www.ncbi.nlm.nih.gov/blast/producttable.shtml BLAST Program Selection Guide at the NCBI]<br> | ||
| + | *[http://www.ncbi.nlm.nih.gov/BLAST/blastcgihelp.shtml Web '''BLAST options page''']<br> | ||
| + | *[http://www.ncbi.nlm.nih.gov/BLAST/tutorial/Altschul-1.html The Statistics of Sequence Similarity Scores]<br> | ||
| + | *[http://bioinformatics.oxfordjournals.org/cgi/content/abstract/18/3/440 '''Pattern Hunter algorithm''']<br> | ||
| + | *[http://prodata.swmed.edu/compass/compass.php '''COMPASS''' on the Web]<br> | ||
| + | |||
| + | |||
| + | |||
| + | <br> | ||
| + | <div style="padding: 10 px; background: #B0B8D7; border:solid 1px #AAAAAA;"> | ||
| + | ====Exercises==== | ||
| + | </div><br> | ||
| + | * In all likelihood, BLAST will be the single most important program you will use for the computational aspects of your work. It is therefore paramount to use it correctly and to understand how and why to set it's parameters and what the output means. Carefully read the [http://www.ncbi.nlm.nih.gov/BLAST/blastcgihelp.shtml '''BLAST Help page''']!<br> | ||
| + | * Read about [http://nar.oxfordjournals.org/cgi/content/full/34/suppl_2/W6 '''BLAST improvements'''] in the 2006 NAR Web server special issue.<br> | ||
| + | * Study the [http://www.ncbi.nlm.nih.gov/blast/producttable.shtml BLAST Program Selection Guide at the NCBI]<br> | ||
| + | * Read about the computational foundations of BLAST in the NCBI tutorials on [http://www.ncbi.nlm.nih.gov/BLAST/tutorial/Altschul-1.html The Statistics of Sequence Similarity Scores], [http://www.ncbi.nlm.nih.gov/BLAST/tutorial/Altschul-2.html Iterated profile searches with PSI-BLAST] and [http://www.ncbi.nlm.nih.gov/BLAST/tutorial/Altschul-3.html The Statistics of PSI-BLAST scores].<br> | ||
| + | * BLAST can be downloaded and installed locally: read the [http://www.ncbi.nlm.nih.gov/blast/download.shtml BLAST download page]. | ||
| + | |||
| + | |||
| + | <br> | ||
| + | <div style="padding: 10 px; background: #879BFA; border:solid 1px #AAAAAA;"> | ||
| + | ==Lecture slides== | ||
| + | </div><br> | ||
| + | <br> | ||
| + | |||
| − | + | <br> | |
| − | ==== | + | <br> |
| − | + | <br> | |
| + | <div style="padding: 10 px; background: #B0B8D7; border:solid 1px #AAAAAA;"> | ||
| + | ===BLAST (Basic Local Alignment Search Tool)=== | ||
| + | </div><br> | ||
| + | <br> | ||
| − | |||
======Slide 004====== | ======Slide 004====== | ||
| − | [[Image: | + | [[Image:08_slide004.jpg|frame|none|Lecture 08, Slide 004<br> |
]] | ]] | ||
======Slide 005====== | ======Slide 005====== | ||
| − | [[Image: | + | [[Image:08_slide005.jpg|frame|none|Lecture 08, Slide 005<br> |
]] | ]] | ||
======Slide 006====== | ======Slide 006====== | ||
| − | [[Image: | + | [[Image:08_slide006.jpg|frame|none|Lecture 08, Slide 006<br> |
| − | + | The [http://www.ncbi.nlm.nih.gov/BLAST/ NCBI '''BLAST''' home page] offers a number of different BLAST "flavours". | |
]] | ]] | ||
======Slide 007====== | ======Slide 007====== | ||
| − | [[Image: | + | [[Image:08_slide007.jpg|frame|none|Lecture 08, Slide 007<br> |
]] | ]] | ||
======Slide 008====== | ======Slide 008====== | ||
| − | [[Image: | + | [[Image:08_slide008.jpg|frame|none|Lecture 08, Slide 008<br> |
| − | + | The enormous speed-up of BLAST is due to its use of an '''indexed table''' of database "words". The index is a list of positions at which each word occurs in the database. Using an index, it is very easy to examine every occurrence of a word in the database and try to extend the word match on both sides with additional similar sequence. The extension does not introduce gaps, because this is faster, but also because the statistics of ungapped alignments are tractable! The final step is the assenbly of significant hits into longer alignments. See also [http://www.ncbi.nlm.nih.gov/sites/entrez?Db=pubmed&Cmd=ShowDetailView&TermToSearch=2231712 Altschul ''et al.'' (1990)]. | |
]] | ]] | ||
======Slide 009====== | ======Slide 009====== | ||
| − | [[Image: | + | [[Image:08_slide009.jpg|frame|none|Lecture 08, Slide 009<br> |
| − | + | Extensive help is available (and should be read!) for each of the options. Take the time to read the [http://www.ncbi.nlm.nih.gov/BLAST/blastcgihelp.shtml '''Web BLAST options document'''] and be sure to understand how to format input, what databases are available and how the choice of database influences the results. If you are not confident with the document, ask on the course list. | |
| − | |||
| − | |||
| − | [ | ||
| − | |||
]] | ]] | ||
======Slide 011====== | ======Slide 011====== | ||
| − | [[Image: | + | [[Image:08_slide011.jpg|frame|none|Lecture 08, Slide 011<br> |
| − | + | Extensive help is available (and should be read!) for each of the options. Be sure to understand the choices and their consequences for [http://www.ncbi.nlm.nih.gov/BLAST/blastcgihelp.shtml#compositional_adjustmentl '''Composition-based statistics'''] and for [http://www.ncbi.nlm.nih.gov/BLAST/blastcgihelp.shtml#filter '''Filtering and Masking'''] segments of low complexity in your query. Filtering is an important option to consider especially for PSI-BLAST searches! | |
]] | ]] | ||
======Slide 012====== | ======Slide 012====== | ||
| − | [[Image: | + | [[Image:08_slide012.jpg|frame|none|Lecture 08, Slide 012<br> |
| − | + | Each Blast "'''hit'''" represents an alignment that can contain one or more HSPs. | |
]] | ]] | ||
======Slide 013====== | ======Slide 013====== | ||
| − | [[Image: | + | [[Image:08_slide013.jpg|frame|none|Lecture 08, Slide 013<br> |
| − | + | Normally scores depend on the matrix that was used and can't be compared between differnet matrices and scoring systems. However the NCBI matrices have been normalized in bits, thus the scores between alignments with different matrices '''can''' be compared, (this is not generally the case with other matrices). In addition the percentage of Identical and similar ("positives") residues and the gap fraction are given. %-Identities and gap fraction are often used to conclude whether two sequences are homologous, the percentage of positives is not usually used since it depends on the matrix. | |
]] | ]] | ||
======Slide 014====== | ======Slide 014====== | ||
| − | [[Image: | + | [[Image:08_slide014.jpg|frame|none|Lecture 08, Slide 014<br> |
| − | + | The E-value is a statistically well founded metric that allows us to conclude the likelihood of a spurious alignment. Computing E-values is possible for HSPs since the statistics of gap-less alignments are analytically tractable, whereas gapped alignments have no theoretical description of the distribution of expected scores.<br> | |
| + | <br> | ||
| + | Note that E-values do not represent an assertion about the retrieved sequence, but an assertion about the score and its relation to the expected distribution of scores. Or, to rephrase this, a large E-value does not mean that your hit is not a homologue, but it means that an irrelevant sequence has a a high chance of having just as high a score due to chance similarities. To repeat: a large E-value does not mean your hit is not a homologue. However a small E-value does indeed mean that a chance alignment is unlikely.<br> | ||
| + | <br> | ||
| + | It is important to realize that the E-value depends on the database size. Obviously, you would expect randomly high-scoring hits more often in a large database than in a small one. Thus an alignment with the '''same score''' will have '''smaller E-value''' searched against a particular genome than if you search it against the entire "nr" dataset of GenBank. (More detail in the NCBI tutorial: [http://www.ncbi.nlm.nih.gov/BLAST/tutorial/Altschul-1.html The Statistics of Sequence Similarity Scores].) | ||
]] | ]] | ||
======Slide 015====== | ======Slide 015====== | ||
| − | [[Image: | + | [[Image:08_slide015.jpg|frame|none|Lecture 08, Slide 015<br> |
| − | + | In the example above, the BLAST search of a Pea defensin - PDB structure 1JKZ - achieved an E-value of only 6.7. However the hit that was retrieved<br> | |
| + | <br> | ||
| + | * is annotated as an ''arabidopsis'' defensin<br> | ||
| + | * has 30% identity over the entire domain, albeit the domain is small<br> | ||
| + | * requires only one single gap for alignment<br> | ||
| + | * and '''has each and every single cysteine conserved, when compared to the query'''!<br> | ||
| + | <br> | ||
| + | Each of these additional observations alone could have led you to conclude homology. The large E-value is primarily due to the fact that the protein sequences are quite short. | ||
]] | ]] | ||
======Slide 016====== | ======Slide 016====== | ||
| − | [[Image: | + | [[Image:08_slide016.jpg|frame|none|Lecture 08, Slide 016<br> |
| − | + | How can there be too many hits, when lots-of-hits is what you are looking for? Either you find redundant sequences or trivially similar sequences that are obscurig the rare, interesting similarities you are looking for (GFP or other fusion proteins come to mind, for example), or you are searching in a database section that contains redundant sequences.<br> | |
| + | <br> | ||
| + | Note that restricting by organism does not restrict the search, but only the list of results that are being reported. The search takes just as long. Only the specialized genome search pages and some non-NCBI databases of model-organism genome projects offer BLAST searches on reduced datasets. These searches are faster. | ||
]] | ]] | ||
======Slide 017====== | ======Slide 017====== | ||
| − | [[Image: | + | [[Image:08_slide017.jpg|frame|none|Lecture 08, Slide 017<br> |
| − | + | How many genes have no homologues? That depends. Unknown genes (or "ORFans") may comprise a significant (albeit diminishing) fraction of genomes. See [http://www.ncbi.nlm.nih.gov/sites/entrez?Db=pubmed&Cmd=ShowDetailView&TermToSearch=12517334 Siew&Fischer (2003)] and a discussion of the role of viral horizontal gene transfer in ORFans by [http://www.biomedcentral.com/1471-2148/6/63 Yin and Fischer (2006)]. In general, between 10 and 30% of sequences may fall into this category and it is likely that even the most closely related species have sequences that are unique. | |
]] | ]] | ||
======Slide 018====== | ======Slide 018====== | ||
| − | [[Image: | + | [[Image:08_slide018.jpg|frame|none|Lecture 08, Slide 018<br> |
]] | ]] | ||
======Slide 019====== | ======Slide 019====== | ||
| − | [[Image: | + | [[Image:08_slide019.jpg|frame|none|Lecture 08, Slide 019<br> |
]] | ]] | ||
======Slide 020====== | ======Slide 020====== | ||
| − | [[Image: | + | [[Image:08_slide020.jpg|frame|none|Lecture 08, Slide 020<br> |
| − | + | Initiate a PSI-BLAST search simply by choosing the option on the BLAST input form. | |
]] | ]] | ||
======Slide 021====== | ======Slide 021====== | ||
| − | [[Image: | + | [[Image:08_slide021.jpg|frame|none|Lecture 08, Slide 021<br> |
]] | ]] | ||
======Slide 022====== | ======Slide 022====== | ||
| − | [[Image: | + | [[Image:08_slide022.jpg|frame|none|Lecture 08, Slide 022<br> |
| − | + | In this example, we are observing how the alignment and score for '''one''' hit from the entire set evolves over a number of iterations. The first E-value is '''<tt>2e-04</tt>'''. | |
]] | ]] | ||
======Slide 023====== | ======Slide 023====== | ||
| − | [[Image: | + | [[Image:08_slide023.jpg|frame|none|Lecture 08, Slide 023<br> |
| − | + | The second E-value for the pair has decreased from <tt>2e-04</tt> to '''<tt>2e-32</tt>'''. This has transformed a somewhat borderline hit to a certain homologue! If you look carefully, you will see that the detailed position of gaps has changed - just like in MSAs, consensus information can be invaluable to place gaps correctly - and the lenght of the alignment has grown considerably. | |
]] | ]] | ||
======Slide 024====== | ======Slide 024====== | ||
| − | [[Image: | + | [[Image:08_slide024.jpg|frame|none|Lecture 08, Slide 024<br> |
]] | ]] | ||
======Slide 025====== | ======Slide 025====== | ||
| − | [[Image: | + | [[Image:08_slide025.jpg|frame|none|Lecture 08, Slide 025<br> |
| − | + | The E-value decreases further. A careful comparison of the trend of E-values can be very helpful for evaluating borderline hits. E-values of homologues almost always get dramatically smaller through the iterations. E-values of spurious hits get larger or stay approximately the same. Make it a habit to look at the '''E-value trend '''in questionable cases '''but exclude the questionable hit from the profile''' by unchecking the check-box on the search form, until you are satisfied that the sequence is a homologue after all. Getting unrelated sequences included in your profile will lead to '''profile corruption'''! | |
]] | ]] | ||
======Slide 026====== | ======Slide 026====== | ||
| − | [[Image: | + | [[Image:08_slide026.jpg|frame|none|Lecture 08, Slide 026<br> |
]] | ]] | ||
======Slide 027====== | ======Slide 027====== | ||
| − | [[Image: | + | [[Image:08_slide027.jpg|frame|none|Lecture 08, Slide 027<br> |
| − | + | In the end, how many false positives can we expect? Unfortunately, more than we'd think. [http://www.ncbi.nlm.nih.gov/sites/entrez?Db=pubmed&Cmd=ShowDetailView&TermToSearch=11893514 Jones & Swindells (2002)] have run an analysis against decoy sequences that picked up false positives in 5% of all cases, after the fifth iteration, although the E-value threshold was set to 0.001. Even though their methodology was a bit ''ad hoc'' and finding false positives about 50 times more frequently than expected is not catastrophic, we must realize that protein sequences are not random strings and that rigorous statistics are very difficult for this complex problem. Use caution, use common sense and in questionable cases try to use independent confirmation of homology, such as conserved binding sites or functional motifs, if possible. | |
]] | ]] | ||
======Slide 028====== | ======Slide 028====== | ||
| − | [[Image: | + | [[Image:08_slide028.jpg|frame|none|Lecture 08, Slide 028<br> |
]] | ]] | ||
| − | |||
| − | |||
| − | + | ||
| + | <br> | ||
| + | <br> | ||
| + | <br> | ||
| + | <div style="padding: 10 px; background: #B0B8D7; border:solid 1px #AAAAAA;"> | ||
| + | ===Other BLAST variations=== | ||
| + | </div><br> | ||
| + | <br> | ||
| + | |||
======Slide 030====== | ======Slide 030====== | ||
| − | [[Image: | + | [[Image:08_slide030.jpg|frame|none|Lecture 08, Slide 030<br> |
| − | + | A nice extension of normal sequence alignment is the graphical view of similarities. But note that BLAST is not an '''optimal''' sequence alignment algorithm and I question why one would use an inferior algorithm if one has better alternatives easily available? Use EMBOSS ''needle'' respectively ''water'' instead! | |
]] | ]] | ||
======Slide 031====== | ======Slide 031====== | ||
| − | [[Image: | + | [[Image:08_slide031.jpg|frame|none|Lecture 08, Slide 031<br> |
]] | ]] | ||
| − | |||
| − | |||
| − | + | ||
| + | <br> | ||
| + | <br> | ||
| + | <br> | ||
| + | <div style="padding: 10 px; background: #B0B8D7; border:solid 1px #AAAAAA;"> | ||
| + | ===Beyond BLAST=== | ||
| + | </div><br> | ||
| + | <br> | ||
| + | |||
======Slide 033====== | ======Slide 033====== | ||
| − | [[Image: | + | [[Image:08_slide033.jpg|frame|none|Lecture 08, Slide 033<br> |
| − | + | Is it possible to improve significantly on BLAST? Yes! An adaptation of the basic strategy of the algorithm improves both the speed and the sensitivity. The Ontario company [http://www.bioinformaticssolutions.com Bioinformatics Solutions] is marketing the [http://bioinformatics.oxfordjournals.org/cgi/content/abstract/18/3/440 '''Pattern Hunter algorithm'''], originally developed by Bin Ma of London and Ming Li of Waterloo.<br> | |
| + | <br> | ||
| + | Besides this being an interesting algorithm, this is an interesting spotlight on the Bioinformatics industry as well. A free academic license is offered for Windows installations only; most "real" bioinformatics would run on some flavor of UNIX machines. And while the fee for the full Academic License is not high (on the order of $1,000.00), the company reports "hundreds" of installed users, in contrast to the tens of thousands who use NCBI BLAST. We note that an important resource in world-wide, daily use does not perform as well as it could, because the resource provider does not acquire the intellectual property of those who could improve it. And since BLAST runs as well as the provider needs to make it to maintain its near monopoly in the user community, there seems to be no incentive for the NCBI to update their servers with PatternHunter. This is clearly the opposite of a win-win situation. What happens in Bionformatics is determined by politics and economics as much as in any other field. | ||
]] | ]] | ||
======Slide 034====== | ======Slide 034====== | ||
| − | [[Image: | + | [[Image:08_slide034.jpg|frame|none|Lecture 08, Slide 034<br> |
| − | + | Why is PatternHunter better? Simply because it uses a more advanced way of defining the database words, or "seeds", that are used to find the initial high-scoring hits. PatternHunter uses '''spaced seeds''', i.e. non-consecutive characters that increase the '''signal to noise''' ratio of similarity, as explained above. Thus the algorithm is both faster (because it spends less time looking at initial seeds that can't be extended well) and more sensitive, because once a hit is accepted, it is more likely to be true. | |
]] | ]] | ||
======Slide 035====== | ======Slide 035====== | ||
| − | [[Image: | + | [[Image:08_slide035.jpg|frame|none|Lecture 08, Slide 035<br> |
| − | + | Is it possible to improve significantly on PSI-BLAST? Yes, [http://www.ncbi.nlm.nih.gov/sites/entrez?cmd=Retrieve&db=pubmed&dopt=AbstractPlus&list_uids=12547212 '''COMPASS''' (Sadreyev & Grishin, 2003)] takes the idea of profile based searches further by aligning profiles of sequences against a database of profiles. The principle is the same as the "equivalence principle" for homology, sometimes we can detect distantly related homologues through a mutual similarity to an intermediate sequence. Run [http://prodata.swmed.edu/compass/compass.php '''COMPASS''' on the Web] against the SCOP database of structural domains (see also here [http://nar.oxfordjournals.org/cgi/content/full/35/suppl_2/W653 Sadreyev ''et al.'' 2007, NAR Web server issue]). | |
| − | |||
| − | ==== | ||
| − | [[ | ||
| − | |||
]] | ]] | ||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | + | <br> | |
| − | + | <br> | |
| − | + | ---- | |
| − | + | <small>[[Lecture_07|(Previous lecture)]] ... [[Lecture_09|(Next lecture)]]</small> | |
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | [[ | ||
| − | |||
| − | ]] | ||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | |||
| − | [[ | ||
| − | |||
| − | ]] | ||
| − | |||
| − | |||
| − | |||
| − | |||
Latest revision as of 03:52, 5 October 2007
(Previous lecture) ... (Next lecture)
Fast Sequence Database Searches
Objectives for this part of the course
- Understand the advantages and limitations of heuristic, local alignment vs. optimal alignment.
- Initiate a BLAST search.
- Understand different BLAST algorithms and for which computational task they are appropriate.
- Understand the contents of the different databases offered by the NCBI for BLASTing and be able to restrict a search by database and organism.
- Understand how to set the algorithm's parameters for different purposes.
- Understand all information in a BLAST report.
- Be able to evaluate the significance of hits through E-values and other metrics / features of the alignment.
- Be able to use PSI-BLAST and avoid and recognize profile corruption; be able to evaluate E-value trends of questionable alignments.
- Be familiar with novel developments beyond BLAST.
Links summary
- BLAST
- BLAST Program Selection Guide at the NCBI
- Web BLAST options page
- The Statistics of Sequence Similarity Scores
- Pattern Hunter algorithm
- COMPASS on the Web
Exercises
- In all likelihood, BLAST will be the single most important program you will use for the computational aspects of your work. It is therefore paramount to use it correctly and to understand how and why to set it's parameters and what the output means. Carefully read the BLAST Help page!
- Read about BLAST improvements in the 2006 NAR Web server special issue.
- Study the BLAST Program Selection Guide at the NCBI
- Read about the computational foundations of BLAST in the NCBI tutorials on The Statistics of Sequence Similarity Scores, Iterated profile searches with PSI-BLAST and The Statistics of PSI-BLAST scores.
- BLAST can be downloaded and installed locally: read the BLAST download page.
Lecture slides
BLAST (Basic Local Alignment Search Tool)
Slide 004

Lecture 08, Slide 004
Slide 005

Lecture 08, Slide 005
Slide 006

Lecture 08, Slide 006
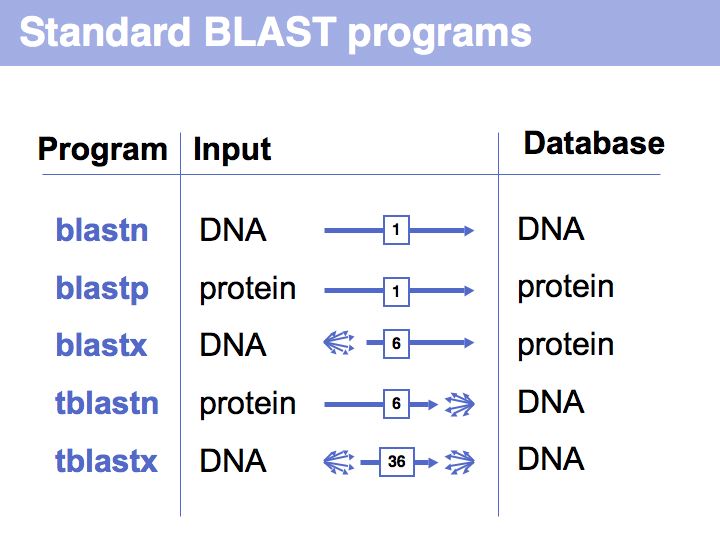
The NCBI BLAST home page offers a number of different BLAST "flavours".
The NCBI BLAST home page offers a number of different BLAST "flavours".
Slide 007

Lecture 08, Slide 007
Slide 008

Lecture 08, Slide 008
The enormous speed-up of BLAST is due to its use of an indexed table of database "words". The index is a list of positions at which each word occurs in the database. Using an index, it is very easy to examine every occurrence of a word in the database and try to extend the word match on both sides with additional similar sequence. The extension does not introduce gaps, because this is faster, but also because the statistics of ungapped alignments are tractable! The final step is the assenbly of significant hits into longer alignments. See also Altschul et al. (1990).
The enormous speed-up of BLAST is due to its use of an indexed table of database "words". The index is a list of positions at which each word occurs in the database. Using an index, it is very easy to examine every occurrence of a word in the database and try to extend the word match on both sides with additional similar sequence. The extension does not introduce gaps, because this is faster, but also because the statistics of ungapped alignments are tractable! The final step is the assenbly of significant hits into longer alignments. See also Altschul et al. (1990).
Slide 009

Lecture 08, Slide 009
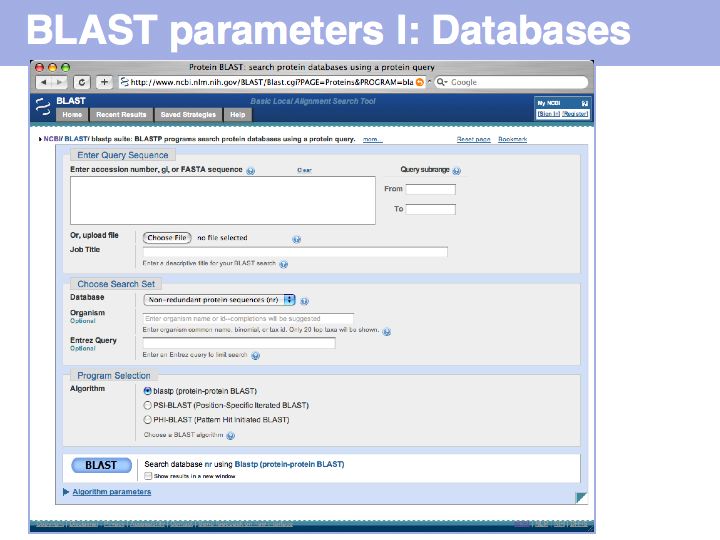
Extensive help is available (and should be read!) for each of the options. Take the time to read the Web BLAST options document and be sure to understand how to format input, what databases are available and how the choice of database influences the results. If you are not confident with the document, ask on the course list.
Extensive help is available (and should be read!) for each of the options. Take the time to read the Web BLAST options document and be sure to understand how to format input, what databases are available and how the choice of database influences the results. If you are not confident with the document, ask on the course list.
Slide 011

Lecture 08, Slide 011
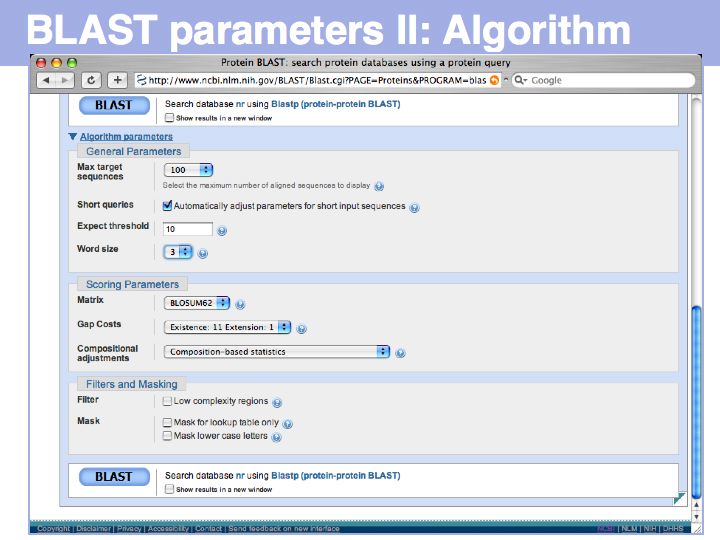
Extensive help is available (and should be read!) for each of the options. Be sure to understand the choices and their consequences for Composition-based statistics and for Filtering and Masking segments of low complexity in your query. Filtering is an important option to consider especially for PSI-BLAST searches!
Extensive help is available (and should be read!) for each of the options. Be sure to understand the choices and their consequences for Composition-based statistics and for Filtering and Masking segments of low complexity in your query. Filtering is an important option to consider especially for PSI-BLAST searches!
Slide 012

Lecture 08, Slide 012
Each Blast "hit" represents an alignment that can contain one or more HSPs.
Each Blast "hit" represents an alignment that can contain one or more HSPs.
Slide 013

Lecture 08, Slide 013
Normally scores depend on the matrix that was used and can't be compared between differnet matrices and scoring systems. However the NCBI matrices have been normalized in bits, thus the scores between alignments with different matrices can be compared, (this is not generally the case with other matrices). In addition the percentage of Identical and similar ("positives") residues and the gap fraction are given. %-Identities and gap fraction are often used to conclude whether two sequences are homologous, the percentage of positives is not usually used since it depends on the matrix.
Normally scores depend on the matrix that was used and can't be compared between differnet matrices and scoring systems. However the NCBI matrices have been normalized in bits, thus the scores between alignments with different matrices can be compared, (this is not generally the case with other matrices). In addition the percentage of Identical and similar ("positives") residues and the gap fraction are given. %-Identities and gap fraction are often used to conclude whether two sequences are homologous, the percentage of positives is not usually used since it depends on the matrix.
Slide 014

Lecture 08, Slide 014
The E-value is a statistically well founded metric that allows us to conclude the likelihood of a spurious alignment. Computing E-values is possible for HSPs since the statistics of gap-less alignments are analytically tractable, whereas gapped alignments have no theoretical description of the distribution of expected scores.
Note that E-values do not represent an assertion about the retrieved sequence, but an assertion about the score and its relation to the expected distribution of scores. Or, to rephrase this, a large E-value does not mean that your hit is not a homologue, but it means that an irrelevant sequence has a a high chance of having just as high a score due to chance similarities. To repeat: a large E-value does not mean your hit is not a homologue. However a small E-value does indeed mean that a chance alignment is unlikely.
It is important to realize that the E-value depends on the database size. Obviously, you would expect randomly high-scoring hits more often in a large database than in a small one. Thus an alignment with the same score will have smaller E-value searched against a particular genome than if you search it against the entire "nr" dataset of GenBank. (More detail in the NCBI tutorial: The Statistics of Sequence Similarity Scores.)
The E-value is a statistically well founded metric that allows us to conclude the likelihood of a spurious alignment. Computing E-values is possible for HSPs since the statistics of gap-less alignments are analytically tractable, whereas gapped alignments have no theoretical description of the distribution of expected scores.
Note that E-values do not represent an assertion about the retrieved sequence, but an assertion about the score and its relation to the expected distribution of scores. Or, to rephrase this, a large E-value does not mean that your hit is not a homologue, but it means that an irrelevant sequence has a a high chance of having just as high a score due to chance similarities. To repeat: a large E-value does not mean your hit is not a homologue. However a small E-value does indeed mean that a chance alignment is unlikely.
It is important to realize that the E-value depends on the database size. Obviously, you would expect randomly high-scoring hits more often in a large database than in a small one. Thus an alignment with the same score will have smaller E-value searched against a particular genome than if you search it against the entire "nr" dataset of GenBank. (More detail in the NCBI tutorial: The Statistics of Sequence Similarity Scores.)
Slide 015

Lecture 08, Slide 015
In the example above, the BLAST search of a Pea defensin - PDB structure 1JKZ - achieved an E-value of only 6.7. However the hit that was retrieved
* is annotated as an arabidopsis defensin
* has 30% identity over the entire domain, albeit the domain is small
* requires only one single gap for alignment
* and has each and every single cysteine conserved, when compared to the query!
Each of these additional observations alone could have led you to conclude homology. The large E-value is primarily due to the fact that the protein sequences are quite short.
In the example above, the BLAST search of a Pea defensin - PDB structure 1JKZ - achieved an E-value of only 6.7. However the hit that was retrieved
* is annotated as an arabidopsis defensin
* has 30% identity over the entire domain, albeit the domain is small
* requires only one single gap for alignment
* and has each and every single cysteine conserved, when compared to the query!
Each of these additional observations alone could have led you to conclude homology. The large E-value is primarily due to the fact that the protein sequences are quite short.
Slide 016

Lecture 08, Slide 016
How can there be too many hits, when lots-of-hits is what you are looking for? Either you find redundant sequences or trivially similar sequences that are obscurig the rare, interesting similarities you are looking for (GFP or other fusion proteins come to mind, for example), or you are searching in a database section that contains redundant sequences.
Note that restricting by organism does not restrict the search, but only the list of results that are being reported. The search takes just as long. Only the specialized genome search pages and some non-NCBI databases of model-organism genome projects offer BLAST searches on reduced datasets. These searches are faster.
How can there be too many hits, when lots-of-hits is what you are looking for? Either you find redundant sequences or trivially similar sequences that are obscurig the rare, interesting similarities you are looking for (GFP or other fusion proteins come to mind, for example), or you are searching in a database section that contains redundant sequences.
Note that restricting by organism does not restrict the search, but only the list of results that are being reported. The search takes just as long. Only the specialized genome search pages and some non-NCBI databases of model-organism genome projects offer BLAST searches on reduced datasets. These searches are faster.
Slide 017

Lecture 08, Slide 017
How many genes have no homologues? That depends. Unknown genes (or "ORFans") may comprise a significant (albeit diminishing) fraction of genomes. See Siew&Fischer (2003) and a discussion of the role of viral horizontal gene transfer in ORFans by Yin and Fischer (2006). In general, between 10 and 30% of sequences may fall into this category and it is likely that even the most closely related species have sequences that are unique.
How many genes have no homologues? That depends. Unknown genes (or "ORFans") may comprise a significant (albeit diminishing) fraction of genomes. See Siew&Fischer (2003) and a discussion of the role of viral horizontal gene transfer in ORFans by Yin and Fischer (2006). In general, between 10 and 30% of sequences may fall into this category and it is likely that even the most closely related species have sequences that are unique.
Slide 018

Lecture 08, Slide 018
Slide 019

Lecture 08, Slide 019
Slide 020

Lecture 08, Slide 020
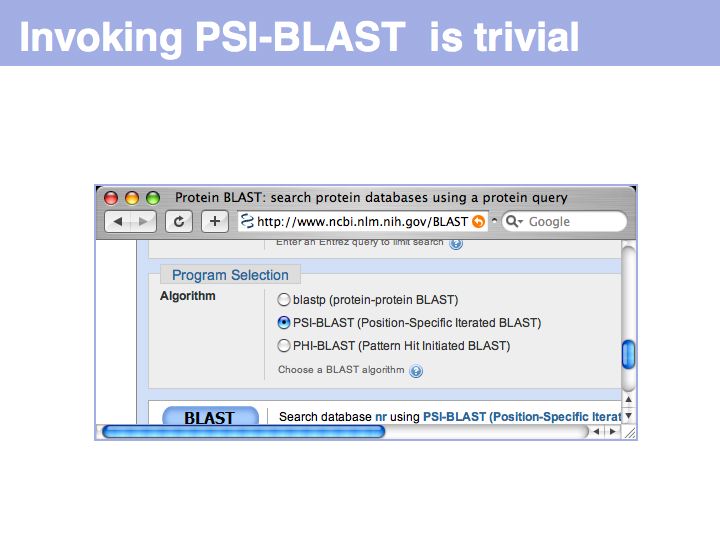
Initiate a PSI-BLAST search simply by choosing the option on the BLAST input form.
Initiate a PSI-BLAST search simply by choosing the option on the BLAST input form.
Slide 021

Lecture 08, Slide 021
Slide 022

Lecture 08, Slide 022
In this example, we are observing how the alignment and score for one hit from the entire set evolves over a number of iterations. The first E-value is 2e-04.
In this example, we are observing how the alignment and score for one hit from the entire set evolves over a number of iterations. The first E-value is 2e-04.
Slide 023

Lecture 08, Slide 023
The second E-value for the pair has decreased from 2e-04 to 2e-32. This has transformed a somewhat borderline hit to a certain homologue! If you look carefully, you will see that the detailed position of gaps has changed - just like in MSAs, consensus information can be invaluable to place gaps correctly - and the lenght of the alignment has grown considerably.
The second E-value for the pair has decreased from 2e-04 to 2e-32. This has transformed a somewhat borderline hit to a certain homologue! If you look carefully, you will see that the detailed position of gaps has changed - just like in MSAs, consensus information can be invaluable to place gaps correctly - and the lenght of the alignment has grown considerably.
Slide 024

Lecture 08, Slide 024
Slide 025

Lecture 08, Slide 025
The E-value decreases further. A careful comparison of the trend of E-values can be very helpful for evaluating borderline hits. E-values of homologues almost always get dramatically smaller through the iterations. E-values of spurious hits get larger or stay approximately the same. Make it a habit to look at the E-value trend in questionable cases but exclude the questionable hit from the profile by unchecking the check-box on the search form, until you are satisfied that the sequence is a homologue after all. Getting unrelated sequences included in your profile will lead to profile corruption!
The E-value decreases further. A careful comparison of the trend of E-values can be very helpful for evaluating borderline hits. E-values of homologues almost always get dramatically smaller through the iterations. E-values of spurious hits get larger or stay approximately the same. Make it a habit to look at the E-value trend in questionable cases but exclude the questionable hit from the profile by unchecking the check-box on the search form, until you are satisfied that the sequence is a homologue after all. Getting unrelated sequences included in your profile will lead to profile corruption!
Slide 026

Lecture 08, Slide 026
Slide 027

Lecture 08, Slide 027
In the end, how many false positives can we expect? Unfortunately, more than we'd think. Jones & Swindells (2002) have run an analysis against decoy sequences that picked up false positives in 5% of all cases, after the fifth iteration, although the E-value threshold was set to 0.001. Even though their methodology was a bit ad hoc and finding false positives about 50 times more frequently than expected is not catastrophic, we must realize that protein sequences are not random strings and that rigorous statistics are very difficult for this complex problem. Use caution, use common sense and in questionable cases try to use independent confirmation of homology, such as conserved binding sites or functional motifs, if possible.
In the end, how many false positives can we expect? Unfortunately, more than we'd think. Jones & Swindells (2002) have run an analysis against decoy sequences that picked up false positives in 5% of all cases, after the fifth iteration, although the E-value threshold was set to 0.001. Even though their methodology was a bit ad hoc and finding false positives about 50 times more frequently than expected is not catastrophic, we must realize that protein sequences are not random strings and that rigorous statistics are very difficult for this complex problem. Use caution, use common sense and in questionable cases try to use independent confirmation of homology, such as conserved binding sites or functional motifs, if possible.
Slide 028

Lecture 08, Slide 028
Other BLAST variations
Slide 030

Lecture 08, Slide 030
A nice extension of normal sequence alignment is the graphical view of similarities. But note that BLAST is not an optimal sequence alignment algorithm and I question why one would use an inferior algorithm if one has better alternatives easily available? Use EMBOSS needle respectively water instead!
A nice extension of normal sequence alignment is the graphical view of similarities. But note that BLAST is not an optimal sequence alignment algorithm and I question why one would use an inferior algorithm if one has better alternatives easily available? Use EMBOSS needle respectively water instead!
Slide 031

Lecture 08, Slide 031
Beyond BLAST
Slide 033

Lecture 08, Slide 033
Is it possible to improve significantly on BLAST? Yes! An adaptation of the basic strategy of the algorithm improves both the speed and the sensitivity. The Ontario company Bioinformatics Solutions is marketing the Pattern Hunter algorithm, originally developed by Bin Ma of London and Ming Li of Waterloo.
Besides this being an interesting algorithm, this is an interesting spotlight on the Bioinformatics industry as well. A free academic license is offered for Windows installations only; most "real" bioinformatics would run on some flavor of UNIX machines. And while the fee for the full Academic License is not high (on the order of $1,000.00), the company reports "hundreds" of installed users, in contrast to the tens of thousands who use NCBI BLAST. We note that an important resource in world-wide, daily use does not perform as well as it could, because the resource provider does not acquire the intellectual property of those who could improve it. And since BLAST runs as well as the provider needs to make it to maintain its near monopoly in the user community, there seems to be no incentive for the NCBI to update their servers with PatternHunter. This is clearly the opposite of a win-win situation. What happens in Bionformatics is determined by politics and economics as much as in any other field.
Is it possible to improve significantly on BLAST? Yes! An adaptation of the basic strategy of the algorithm improves both the speed and the sensitivity. The Ontario company Bioinformatics Solutions is marketing the Pattern Hunter algorithm, originally developed by Bin Ma of London and Ming Li of Waterloo.
Besides this being an interesting algorithm, this is an interesting spotlight on the Bioinformatics industry as well. A free academic license is offered for Windows installations only; most "real" bioinformatics would run on some flavor of UNIX machines. And while the fee for the full Academic License is not high (on the order of $1,000.00), the company reports "hundreds" of installed users, in contrast to the tens of thousands who use NCBI BLAST. We note that an important resource in world-wide, daily use does not perform as well as it could, because the resource provider does not acquire the intellectual property of those who could improve it. And since BLAST runs as well as the provider needs to make it to maintain its near monopoly in the user community, there seems to be no incentive for the NCBI to update their servers with PatternHunter. This is clearly the opposite of a win-win situation. What happens in Bionformatics is determined by politics and economics as much as in any other field.
Slide 034

Lecture 08, Slide 034
Why is PatternHunter better? Simply because it uses a more advanced way of defining the database words, or "seeds", that are used to find the initial high-scoring hits. PatternHunter uses spaced seeds, i.e. non-consecutive characters that increase the signal to noise ratio of similarity, as explained above. Thus the algorithm is both faster (because it spends less time looking at initial seeds that can't be extended well) and more sensitive, because once a hit is accepted, it is more likely to be true.
Why is PatternHunter better? Simply because it uses a more advanced way of defining the database words, or "seeds", that are used to find the initial high-scoring hits. PatternHunter uses spaced seeds, i.e. non-consecutive characters that increase the signal to noise ratio of similarity, as explained above. Thus the algorithm is both faster (because it spends less time looking at initial seeds that can't be extended well) and more sensitive, because once a hit is accepted, it is more likely to be true.
Slide 035

Lecture 08, Slide 035
Is it possible to improve significantly on PSI-BLAST? Yes, COMPASS (Sadreyev & Grishin, 2003) takes the idea of profile based searches further by aligning profiles of sequences against a database of profiles. The principle is the same as the "equivalence principle" for homology, sometimes we can detect distantly related homologues through a mutual similarity to an intermediate sequence. Run COMPASS on the Web against the SCOP database of structural domains (see also here Sadreyev et al. 2007, NAR Web server issue).
Is it possible to improve significantly on PSI-BLAST? Yes, COMPASS (Sadreyev & Grishin, 2003) takes the idea of profile based searches further by aligning profiles of sequences against a database of profiles. The principle is the same as the "equivalence principle" for homology, sometimes we can detect distantly related homologues through a mutual similarity to an intermediate sequence. Run COMPASS on the Web against the SCOP database of structural domains (see also here Sadreyev et al. 2007, NAR Web server issue).