Integrator Unit: Mutation Impact
Boris Steipe
|
Expected Preparations:
|
|||||||||||||||
|
|
|||||||||||||||
| Keywords: Integrator unit: assess the impact of mutations in a gene | |||||||||||||||
|
|
|||||||||||||||
|
Objectives:
|
Outcomes:
|
||||||||||||||
|
|
|||||||||||||||
|
Deliverables: Integrator unit: Deliverables can be submitted for course marks. See below for details. |
|||||||||||||||
|
|
|||||||||||||||
|
Evaluation: Material based on this Integrator Unit can be submitted for summative feedback (course marks). To submit:
|
|||||||||||||||
Contents
This page integrates material from the learning units for R programming, working with sequences and the genetic code, and probability and significance, in a task for evaluation.
Biological Context
Cancer is a genetic disease and one aspect that makes cancer hard to treat is that cancer cells adapt and evolve and as a result of selective pressure from the body’s own defenses, they become progressively more aggressive and treatment-resistant. Since the cancer phenotype is ultimately based on genetic alterations, it is important to understand which genes contribute. Unfortunately this is not as simple as just sequencing a few cancers: one of the hallmarks of the disease is genome instability (this contributes to the accelerated evolution), and it is very difficult to distinguish causal mutations from incidental mutations, or, driver genes from passenger genes.
However, an analysis of the distribution of mutations may help. Passenger mutations are expected to be randomly distributed throughout the genome, driver mutations are expected to have either a gain of function or loss of function effect. Gain of function mutations are expected to be very specific, targeting only a small number of amino acids in a defined region of the protein. We actually expect purifying selection against mutations elsewhere. Loss of function mutations are expected to include nonsense mutations, frameshifts, but above all, they should be enriched in missense and nonsense mutations relative to silent mutations.
The task of this unit is to analyze the relative frequencies of neutral, missense and nonsense mutations in a gene, and contrast that with the frequencies one would expect if the distribution of mutations was purely due to chance. This analysis should work on an actual sequence, and consider actually observed mutations. We will develop it to evaluate mutations of the KRas gene, a known cancer driver, an olfactory receptor (OR1A1), most likely not involved in cancer, and the PTPN11 phosphatase, a gene of interest whose role in cancer we would like to understand better.
KRas and cancer
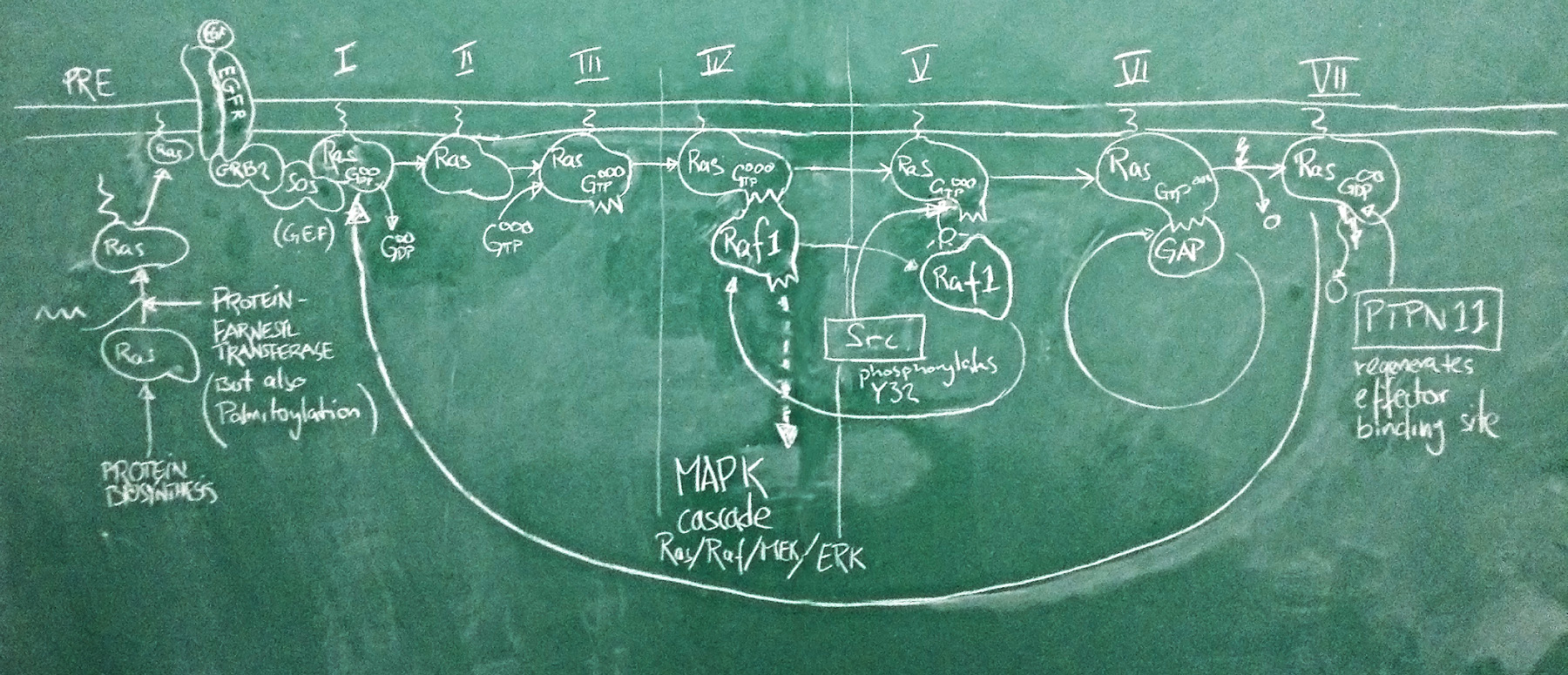
Sketch of the Ras activation cycle. (PRE) Ras is translated, and C-terminally farnesylated, palmitoylated and located to the plasma membrane. (I) The GEF Sos is activated in its complex with active EGFR. It binds to Ras and removes GDP. (II) Apo-Ras is ready to bind a nucleotide. (III) upon GTP binding, Ras acquires its active conformation. (IV) Ras binds its effectors such as Raf1. This switches the MAPK signalling cascade on and leads to cell proliferation. (V) Src phosporylates Ras Y32. This reduces the affinity of RAf1 by ~1000-fold. (VI) GAPs can now displace the effector and stimulate Ras GTPase activity. GTP is hydrolyzed to GDP. (VII) with bound GDP, Ras acquires the inactive conformation. PTPN 11 removes the Y32 phosphate and regenerates the effector binding site. The cycle can begin anew.
Nucleotide binding domains are among the oldest known protein families and one family in particular, the G-proteins, has diverse roles in all domains of life. These are collectively called GTP hydrolases, or GTPases – a misnomer, since even though they do catalyze the hydrolysis of GTP to GDP, their role in the cell has nothing to do with GTP metabolism, but comes from a conformational change that accompanies binding to either GTP or GDP. As far as enzymes go, GTPases are rather slow. They are switches, not gears.
A large family among these G-proteins are the Ras proteins. In humans, there are three isoforms of Ras called HRas, KRas and NRas. These are differentially expressed in tissues and have slightly different C-termini through which they are localized to different membrane subdomains. When Ras binds GTP, it adopts a stable, active ON conformation through which it activates effector proteins. But then the Ras protein slowly hydrolyses GTP to GDP, it undergoes a conformational change and enters the OFF state. Then GDP dissociates from the binding site, Ras can re-bind GTP and is once again switched ON. This cycle is modified by interactors: GEF proteins (Guanine Nucleotide Exchange factors such as Sos) catalyze the dissociation of GDP and thus speed up the re-uptake of GTP and re-activation of Ras. Thus they shift the cycle to an active state. GAP proteins (GTPase activating proteins such as P120GAP) speed up the conversion of GTP to GDP. This shifts the cycle towards its inactive state.
One of the most important pathways for cell proliferation is the EGFR pathway that feeds into the MAPK cascade. Under physiological conditions, the active EGFR activates the Sos protein, which shifts a pool of Ras molecules into their active state. Active Ras then switches its effectors on – among them Raf1 – which activates a signalling cascade that induces cell proliferation. This is limited by GAPs that speed up Ras GTPase activity which turns the Ras OFF again. Deactivation of Sos when the EGFR is inactive ensures that GDP remains bound and the Ras protein pool remains OFF. This matches our expectations about the roles of these proteins well.
The problem is that this system can go terribly wrong if Ras gets mutated in a way that damages its catalytic activity and prevents GTP hydrolysis. Activating GAPs no longer works to switch Ras OFF, because if the Ras active site is dead, GAPs have no way of inducing it. And inhibiting GEFs does not switch Ras OFF either, because GTP does not get hydrolyzed to GDP and there is no need for GEFs to clear the active site of GDP. The switch is ON and stays ON. The EGFR pathway is on and stays on. The cell proliferates out of control. This can be the first step of transforming a cell into a cancer cell and this exact mutation in the KRas protein is the second-most frequent mutation seen in cancer genome studies (behind p53) and possibly the most powerful cancer driver mutation of all. The big issue about all this is that mutant Ras is generally considered “undruggable”:1 we can’t imagine small molecule drugs that would restore Ras’ catalytic activity, and the affinity of GTP to the molecule is so high that we haven’t found competitive antagonists that don’t have dramatic side effects. An interesting new development therefore was the recent discovery that a phosphatase - PTPN11 - somehow works synergistically with Ras to facilitate its activation of effectors: inhibition of PTPN11 suppressed oncogenesis2. If this is a pathophysiologically relevant effect, we expect cancer mutations to spare PTPN11, or even to deregulate it to enhance its activity.
Do they?
Cancer gene data
Knowledge about the mutations of cancer comes from large-scale genome sequencing efforts of cancer tissue samples, and is collected and curated by a small number of databases. These databases sift through the massive volumes of sequence changes, distinguish natural variation from novel somatic mutations, and map the nucleotide changes to individual genes. One of these resources is the IntOGen database in Barcelona.
Task…
- visit IntOGen.
- find the KRas information page and briefly explore the information that is available.
- then visit the information pages for three other genes:
- Rab39B, a small GTPase, homologous to KRAS, but not associated with cancer;
- PTPN11, the non-receptor type protein-phosphatase discussed above;
- PTPN5, a homologous phosphatase that appears not to be associated with cancer.
- Here, Kras and PTPN11 are proteins which need to be studied regarding their role in cancer; Rab39B and PTPN5 serve as controls: they have about the same size and similar domain composition - but are not active in cancer-relevant pathways.
Loading Data
Task…
To prepare your report you first need to load data:
- Open the RStudio course project.
- Begin a new R script to explore KRas, Rab39B, PTPN11 and PTPN5 mutations.
- Start by collecting four FASTA sequences that I have provided in the
data/directory into a data frame. Something like:
myFA <- readFASTA("data/RAB39B_HSa_coding.fa")
myFA <- rbind(myFA, readFASTA("data/PTPN5_HSa_coding.fa"))
myFA <- rbind(myFA, readFASTA("data/PTPN11_HSa_coding.fa"))
myFA <- rbind(myFA, readFASTA("data/KRAS_HSa_coding.fa"))… should work fine. Give your data frame convenient, meaningful row names - do not refer to the rows simply by row index in your script. Gene names will work fine.
For the Report Option…
Task…
- Write code that executes a loop
Ntimes (forN <- 100000) to create a point mutation randomly in a gene.3 Keep track of the number of missense, silent (“synonymous”), and nonsense (“truncating”)” mutations you find. To develop your code, use a smaller size of N, obviously. - Put your code into a function that takes as parameters a single string of nucleotides as input, and the number of trials for your simulation. The number of mutations in eaqch category should be returned in a list.
- There are many ways to handle your nucleotide string input
for this purpose; you need to figure out what works best for you. You
could consider producing a vector of nucleotides and keep track of codon
positions separately, or a matrix with three columns in which every row
is a codon, or you could leave the string as-is and use
substring()to extract codons, or come up with a different solution. Also you could usebiostrings::functions orseqinr::for the translation. Or just use code that is similar to the learning units. Don’t forget to reference your sources! - Tests: validate your function as follows.
- The sequence
ATGATGATGATGATGATGhas no silent mutations; - The sequence
CCCCCCCCCCCCCCCCCChas no truncating mutations; - The sequence
TATTACTATTACTATTAChas some truncations, about the same frequency asTGGTGGTGGTGGTGGTGGTGGTGG; both those sequences have about twice as many truncating mutations asTGTTGTTGTTGTTGTTGTTGTTGT. (Explain what these tests demonstrate.)
- The sequence
- Once your function is ready, run your simulation once for each of the four genes: Kras, Rab39B, PTPN5, and PTPN11. Paste the exact output of each of the four runs into your report.
- For each of the four genes, discuss the relative frequency of the mutations you have observed in each category and compare it to the frequency that you find on the IntOGen Web site.
- Explain whether you think there is an important difference between the expected categories of mutations (i.e. the stochastic background that you simulated), and categories of mutations that were observed in cancer genomes.
- Write a short report that interprets your results against the context of cancer biology outlined above: what would you expect if any of these genes were cancer drivers, what do you observe, what can you conclude from your observation?
Questions, comments
If in doubt, ask! If anything about this contents is not clear to you, do not proceed but ask for clarification. If you have ideas about how to make this material better, let’s hear them. We are aiming to compile a list of FAQs for all learning units, and your contributions will count towards your participation marks.
Improve this page! If you have questions or comments, please post them on the Quercus Discussion board with a subject line that includes the name of the unit.
References
Rhett, J M,
Imran Khan, and John P O’Bryan. (2020). “Biology, pathology, and
therapeutic targeting of RAS”. Advances in Cancer Research
148:69–146 .
[PMID: 32723567]
[DOI: 10.1016/bs.acr.2020.05.002]
Bunda,
Severa et al.. (2015). “Inhibition of SHP2-mediated
dephosphorylation of Ras suppresses oncogenesis”. Nature
Communications 6:8859 .
[PMID: 26617336]
[DOI: 10.1038/ncomms9859]
Kano,
Yoshihito et al.. (2019). “Tyrosyl phosphorylation of KRAS
stalls GTPase cycle via alteration of switch I and II conformation”.
Nature Communications 10(1):224 .
[PMID: 30644389]
[DOI: 10.1038/s41467-018-08115-8]
[END]